Parmi les méthodes à la disposition du chimiste pour l'analyse structurelle, la résonance magnétique nucléaire (RMN) et l'infrarouge (IR) jouent des rôles complémentaires : la RMN permet de connaître l'enchaînement des atomes, l'infrarouge les groupes fonctionnels présents dans les molécules. Même si au cours des dernières décennies la RMN a fait d'énormes progrès, en particulier à l'aide des techniques pulsées à transformée de Fourier, l'infrarouge reste parfois le seul moyen de lever les ambiguïtés pouvant subsister quant à la structure d'une molécule.
Dans cet exposé, nous allons dans un premier temps nous intéresser aux bases physiques de l'infrarouge sur les molécules diatomiques avant de généraliser cette étude aux molécules polyatomiques pour introduire les éléments d'analyse utiles au chimiste organicien, et enfin conclure sur les différents types d'appareillage utilisés.
Vibrations dans les molécules diatomiques
Considérons une molécule diatomique AB et notons r la distance internucléaire. L'énergie potentielle du système est représentée par la courbe suivante (fig. 1), dite courbe de Morse, dont le minimum correspond à la distance internucléaire d'équilibre r0.

Autour de cette position, nous pouvons effectuer un développement limité dont le premier terme non nul sera en (r-r0)2. Ainsi, au voisinage de la position d'équilibre, l'énergie potentielle peut s'exprimer sous la forme :
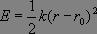
qui correspond à l'énergie d'un oscillateur harmonique, modèle que nous utiliserons pour étudier les vibrations de la liaison A-B.
L'oscillateur harmonique en mécanique classique
Nous nous placerons dans le référentiel barycentrique du système (A, mA), (B, mB), galiléen dès lors que le système est isolé, l'étude peut alors se ramener à celle d'une particule de masse 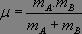 , dite masse réduite, définie par sa position r (égale à la distance internucléaire) par rapport au centre d'inertie.
, dite masse réduite, définie par sa position r (égale à la distance internucléaire) par rapport au centre d'inertie.
Dans ce référentiel, la conservation de l'énergie s'écrit :
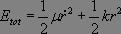
Soit en dérivant par rapport au temps :
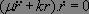
En supposant de plus que le système n'est pas à l'équilibre (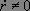 ), nous obtenons l'équation différentielle d'évolution :
), nous obtenons l'équation différentielle d'évolution :
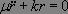
Les solutions sont de la forme :
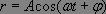
où 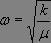 , soit
, soit 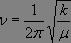 , ce qui constitue la loi de Hooke.
, ce qui constitue la loi de Hooke.
Nous voyons que la fréquence propre du système dépend de la masse réduite µ et de la force de la liaison, comme le montrent les résultats expérimentaux suivants :
| Liaison | Nombre d'onde σ (cm-1) |
|---|---|
| C-H | 3000 |
| C-D | 2100 |
| C-Cl | 700 |
| C≡C | 2200 |
| C=C | 1640 |
| C-C | 1000 |
| C≡O | 2700 |
| C=O | 1715 |
| C-O | 1100 |
| O-H | 3500 |
| N-H | 3300 |
| C-H | 3000 |
Même s'il permet d'expliquer les fréquences d'absorption, le modèle classique ne permet pas de décrire le comportement d'un système soumis à une onde électromagnétique, c'est pourquoi nous allons devoir recourir à une étude quantique, plus pertinente.
L'oscillateur harmonique en mécanique quantique
L'équation de Schrödinger du système précédent s'écrit :
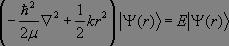
La résolution mathématique est possible et conduit à des énergies propres de la forme :
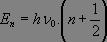
où ν0 est la fréquence d'absorption déterminée précédemment.
Cette expression nous montre que même à 0 K, il existe une énergie et donc un mouvement de vibration. De plus, les énergies mises en jeu en infrarouge (environ 25 kJ.mol-1) comparées à l'agitation thermique (2,5 kJ.mol-1 à 298 K), montrent que seul l'état fondamental de vibration sera peuplé.
Les vecteurs propres associés sont déterminés par la formule suivante :
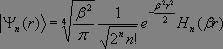
où 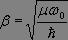 et où Hn(r) représente le polynôme d'Hermite d'ordre n, qui seul nous importera par la suite, dont l'expression est donnée par la relation :
et où Hn(r) représente le polynôme d'Hermite d'ordre n, qui seul nous importera par la suite, dont l'expression est donnée par la relation :
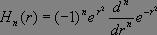
A présent que les énergies et les fonctions propres associées de l'oscillateur harmonique connues, nous allons pouvoir étudier l'interaction entre ce dernier et une onde électromagnétique.
Interaction avec une onde électromagnétique
Une onde électromagnétique est composée d'un champ électrique 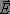 et d'un champ magnétique
et d'un champ magnétique 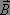 qui interagissent respectivement avec le moment dipolaire
qui interagissent respectivement avec le moment dipolaire 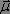 et le moment magnétique
et le moment magnétique 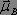 . L'énergie d'interaction est donnée par la formule suivante :
. L'énergie d'interaction est donnée par la formule suivante :
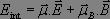
Le terme d'interaction magnétique est négligeable devant le terme d'interaction électrique. Nous en déduisons la forme de l'hamiltonien perturbé par l'onde appliquée :
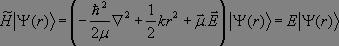
Nous ne traiterons cette perturbation qu'au premier ordre : nous considérerons que les niveaux d'énergie et les états propres de l'oscillateur harmonique ne sont pas modifiés par la perturbation appliquée, cette dernière n'induisant que le passage d'un état vers un autre. Considérons le système dans un état initial Ψi et étudions son passage à l'état Ψj.
La probabilité de passage entre les états i et j est proportionnelle au produit :
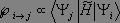
que nous pouvons développer :
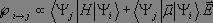
Le premier terme est nul dès lors que i ≠ j puisque les fonctions propres de l'oscillateur harmonique sont orthogonales entre elles, et ce cas ne représente pas une transition. Pour le second terme, nous effectuons un développement limité au premier ordre du moment dipolaire électrique :
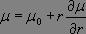
Il vient alors :
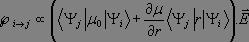
Comme précédemment, le premier terme est nul dès lors que nous observons une transition. Ainsi, pour qu'une transition puisse être observée, il faut que :
-
la vibration modifie la valeur du moment dipolaire électrique
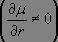 ,
, -
le produit
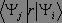 soit non nul. Pour cela, il convient d'expliciter le produit à partir des fonctions propres de l'oscillateur harmonique :
soit non nul. Pour cela, il convient d'expliciter le produit à partir des fonctions propres de l'oscillateur harmonique :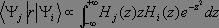
Compte tenu des relations de récurrence vérifiées par les polynômes d'Hermite :
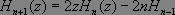
Il vient :
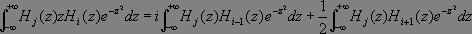
Or :
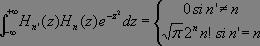
Le premier terme est non nul dès lors que j = i - 1 (émission) et le second si j = i + 1 (absorption).
Ce qui nous montre que la transition n'est possible que si j = i ± 1 et que par conséquent, le quantum d'énergie absorbé ou émis a toujours la seule fréquence ν0, donnée par la loi de Hooke.
Structure fine
En plus du mouvement de vibration, chaque molécule diatomique possède un mouvement de rotation, d'énergie moindre, qui induit l'existence d'une structure fine des transitions. Son étude n'entre pas dans le cadre de cet exposé, mais elle implique une multiplication des raies qui peut devenir une bande si la résolution devient insuffisante. Pour observer cette structure fine, il est nécessaire de travailler en phase gazeuse afin de permettre aux différentes molécules de tourner librement sans changer trop souvent d'état rotationnel lors d'une collision avec une autre molécule (Figure 2).
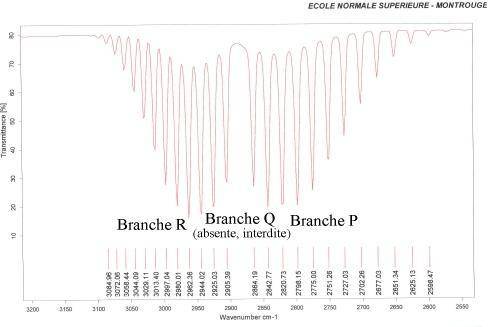
Après avoir établi les différents résultats de spectroscopie infrarouge sur le cas de la molécule diatomique, nous allons généraliser cette étude aux molécules polyatomiques.
Vibrations dans les molécules polyatomiques
Considérons une molécule composée de N atomes. 3N coordonnées sont alors nécessaires pour repérer les différentes positions des atomes. Le mouvement global de cette molécule est repéré par 6 coordonnées (3 pour la translation du centre de gravité, et 3 pour les rotations du système autour de celui-ci) ou seulement 5 dans le cas des molécules linéaires (la rotation autour de l'axe de la molécule n'a pas de signification physique), il reste par conséquent 3N-6 (ou 3N-5 pour les molécules linéaires) variables attribués aux mouvements de vibration.
Cas d'une molécule linéaire : le dioxyde de carbone
Composée de trois atomes, la molécule de dioxyde de carbone (CO2) possède 4 mouvements de vibration indépendants sur lesquels tout mouvement de vibration peut se décomposer. Ces mouvements sont appelés modes propres de vibration.
Ces différents mouvements sont les suivants :

D'un point de vue général, les différents modes sont classés en fonction de deux paramètres :
-
un mode de vibration qui modifie les longueurs des liaisons est dite d'élongation, sinon, c'est une vibration de déformation angulaire (plane ou non),
-
un mode de vibration qui conserve la symétrie moléculaire est dit symétrique (indice s), il est asymétrique s'il conduit à la perte d'un ou plusieurs éléments de symétrie de la molécule.
Parmi les différents modes de vibration du dioxyde de carbone, seuls trois modifient le moment dipolaire électrique de la molécule (dont deux sont dégénérés) et seront donc actifs en infrarouge. En pratique le dioxyde de carbone présente une absorption à 2349 cm-1, souvent observée sur les spectres utilisant l'air comme milieu de référence du fait d'une mauvaise compensation.
Cas d'une molécule non linéaire : la molécule d'eau
La molécule d'eau (H2O) est composée de trois atomes et est de géométrie coudée, elle possède par conséquent 3 modes propres de vibration (Figure 4).
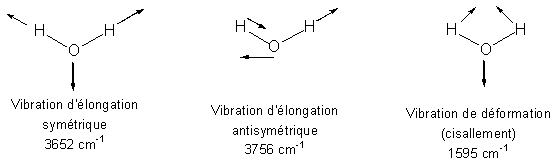
Les différents modes de vibration du groupe méthylène (CH2) ou plus généralement d'un carbone tétraédrique sont similaires, mais du fait de la présence d'autres substituants, il possède six modes propres1 (Figure 5).
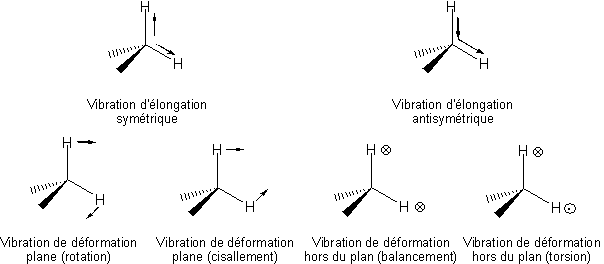
Généralisation
Dès que les molécules deviennent compliquées, des études telles que celles que nous avons présentées ci-dessus ne sont plus aussi simples, et il est souvent impossible d'attribuer toutes les bandes d'un spectre infrarouge. Toutefois, et c'est ce qui fait la force de cette spectroscopie, de nombreux groupements fonctionnels possèdent des bandes d'absorption caractéristiques qui signalent leur présence à l'intérieur d'une molécule.
Le spectre Infrarouge en chimie organique
Pour le chimiste organicien, la spectroscopie infrarouge ne sert plus qu'à la détermination des fonctionnalités présentes dans la molécule, la structure ou les différents fragments de structures, étant principalement déterminée par RMN.
Toutefois, certains signaux ne sont pas forcément observés en RMN, comme c'est le cas des protons échangeables, et c'est là que la spectroscopie infrarouge prend tout son sens.
Les différentes zones du spectre en Infrarouge
D'une manière générale, les masses des atomes étudiés en chimie organique (C, N, O... ) sont quasiment identiques, de même, les énergies des liaisons sont relativement semblables, ce qui conduit à dégager différentes zones du spectre en fonction de l'absorption observée :
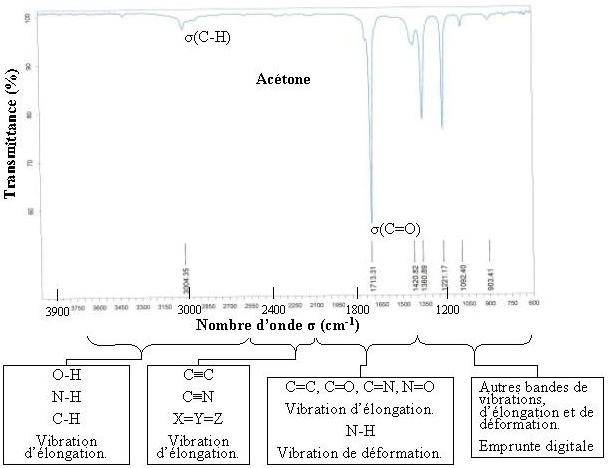
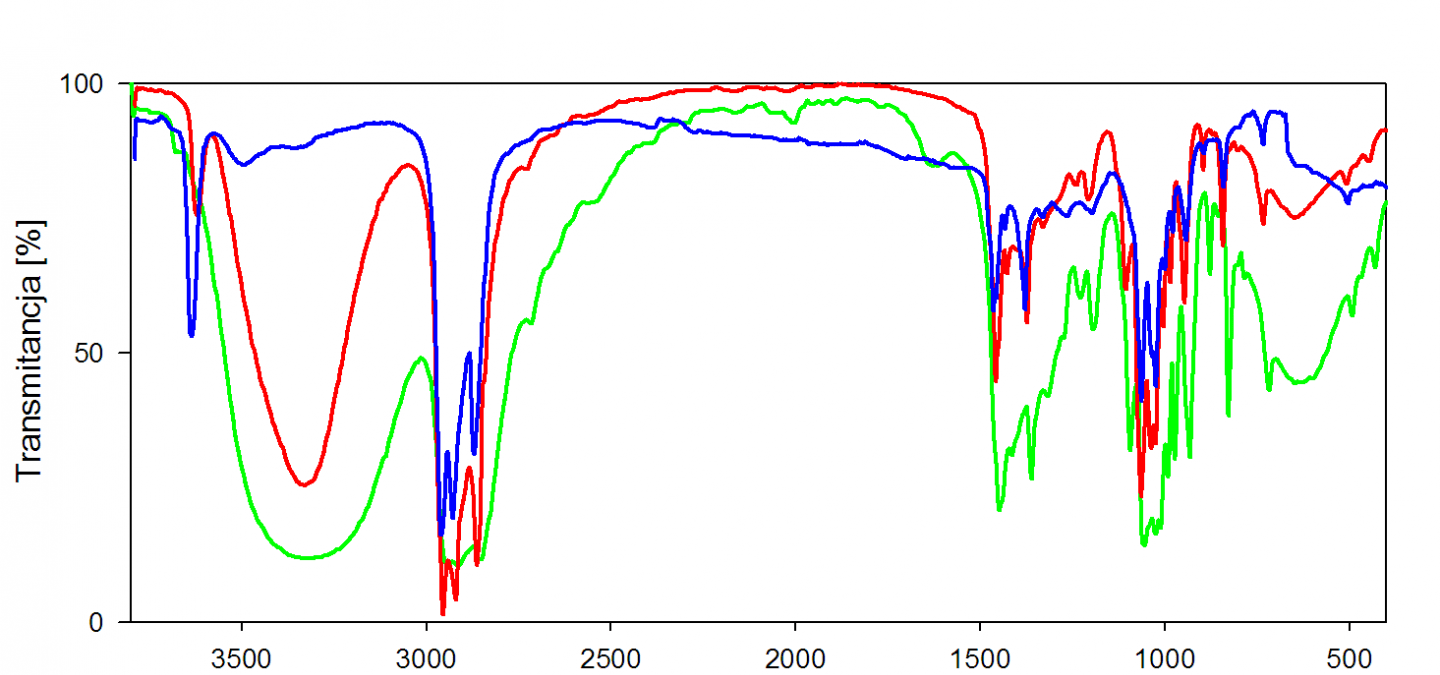
Un spectre infrarouge est traditionnellement présenté en transmittance (fraction de l'intensité transmise par rapport à l'intensité incidente) exprimée en pourcentage et l'axe des abscisses en fonction du nombre d'onde (inverse de la longueur d'onde), sur un axe dirigé vers la gauche.
La loi de Beer-Lambert est vérifiée en infrarouge, ce qui en fait une méthode d'analyse quantitative, il faut toutefois se souvenir que cette loi utilise les absorbances (logarithme de la transmittance) dans son application.
Une analyse plus fine permet alors d'identifier les différentes fonctions présentes.
Les vibrations d'élongation des liaisons X-H
D'après la loi de Hooke, deux paramètres sont à prendre en compte pour l'étude des vibrations d'élongation de la liaison X-H. Le premier est l'atome X (et plus particulièrement sa masse), nous distinguerons le cas des atomes d'oxygène, d'azote et de carbone, ces derniers étant les plus fréquemment rencontrés en chimie organique.
La vibration d'élongation des liaisons O-H et N-H
En phase gazeuse, les vibrations d'élongation des liaisons O-H et N-H se présentent sous la forme d'une bande fine, respectivement entre 3590 et 3650 cm-1 et entre 3300 et 3500 cm-1. En solution ou en liquide pur, les hydrogènes sont engagés dans des liaisons H, ce qui a pour effet, d'une part d'affaiblir la liaison (baisse du nombre d'onde) et d'autre part de provoquer un élargissement des raies par effet de solvatation.
Pendant longtemps, l'infrarouge a permis de quantifier la force des liaisons H, les nombres d'onde de vibration dépendant largement de l'atome avec lequel le pont était engagé :
|
X = O ou NR |
σv(O-H) = 2500 à 3200 cm-1 forte intensité |
|
X = O ou NR2 |
σv(O-H) = 3200 à 3600 cm-1 intensité variable |
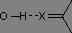
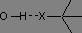
Ainsi, en solution concentrée ou en liquide pur, les alcools ont tendance à présenter une large bande OH entre 3200 et 3600 cm-1 alors que les acides carboxyliques, eux, présenteront une bande beaucoup plus large entre 2500 et 3200 cm-1.
Les hydrogènes des atomes d'azote ayant moins tendance à former des liaisons H que leurs analogues oxygénés, les amines ne présentent pas un tel déplacement. L'infrarouge permet toutefois de distinguer les différentes classes d'amines, puisque le groupe NH2 d'une amine primaire présentera deux bandes de vibration N-H, correspondant aux modes symétriques et antisymétriques, alors qu'une amine secondaire ne présentera qu'une seule bande d'intensité moyenne.Les vibrations d'élongation des liaisons C-H
Les bandes de vibration d'élongation des liaisons C-H s'étendent sur une plage allant de 2850 à 3100 cm-1. Un des intérêts principaux est que les vibrations des liaisons C-H dépendent largement de l'atome de carbone portant l'hydrogène : les hydrogènes liés à un carbone saturé présentent leurs bandes de vibration en deçà de 3000 cm-1, alors que dans le cas d'un carbone insaturé, ces bandes sont au-delà de 3000 cm-1.
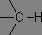 |
σv(C-H) = 2850 à 2960 cm-1 |
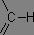 |
σv(C-H) = 3000 à 3100 cm-1 |
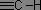 |
σv(C-H) = 3300 cm-1 |
La vibration d'élongation de la liaison C=O
La vibration d'élongation de la liaison C=O absorbe entre 1650 et 1800 cm-1, région peu sollicitée par d'autres absorptions, et est donc facilement reconnaissable. De nombreuses informations peuvent être obtenues à partir de la position de la bande d'absorption.
Composés carbonylés
Les aldéhydes présentent une absorption caractéristique entre 1720 et 1740 cm-1, les cétones entre 1705 et 1725 cm-1. Dès lors que le carbonyle est conjugué, ces valeurs sont abaissées de 15 à 40 cm-1.
La position de la bande d'absorption est aussi largement influencée par la tension de cycle, ce qui permet de caractériser facilement la taille d'un cycle (tendu) comportant un carbonyle.
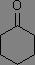 |
σv(C=O) = 1685 à 1705 cm-1 |
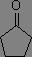 |
σv(C=O) = 1740 à 1750 cm-1 |
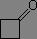 |
σv(C=O) = 1780 cm-1 |
De plus, le carbonyle accepteur de liaison H voit sa fréquence d'absorption diminuer de 40 à 60 cm-1.
Dérivés des acides carboxyliques
Dans les systèmes RC(=O)X, plus X est électronégatif, plus la fréquence d'absorption est augmentée.
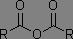 |
1800 à 1850 cm-1 1740 à 1790 cm-1 |
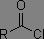 |
1790 à 1815 cm-1 |
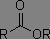 |
1735 à 1750 cm-1 |
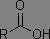 |
1700 à 1725 cm-1 |
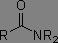 |
1630 à 1700 cm-1 |
Ces bandes sont largement influencées par la présence de liaisons H.
Vibrations de déformation dans les composés aromatiques
Les vibrations de déformation ont en général une fréquence d'absorption plus basse et une intensité plus faible que les vibrations d'élongation, ce qui amène les bandes dans la zone d'empreinte digitale et rend leur repérage et leur analyse délicate.
Toutefois, dans le cas des composés aromatiques, les bandes de déformation hors du plan, situées entre 680 et 900 cm-1, sont intenses et permettent de déterminer les différentes substitutions du cycle aromatique.
|
5H voisins (monosubstitué) |
735 à 770 cm-1 685 à 710 cm-1 forte intensité |
|
4H voisins (o-disubstitué) |
740 à 760 cm-1 forte intensité |
|
3H voisins (m-disubstitué, 1,2,3-trisubstitué) |
770 à 800 cm-1 forte intensité |
|
2H voisins (p-disubstitué...) |
800 à 840 cm-1 forte intensité |
|
H isolé (p-disubstitué...) |
800 à 900 cm-1 intensité faible (souvent non visible) |
Après avoir étudié la théorie de la spectroscopie infrarouge, puis vu quelques éléments d'analyse de spectres, nous nous proposons d'étudier les différentes techniques qui permettent d'acquérir ces spectres.
Techniques Expérimentales en spectroscopie Infrarouge
Deux techniques principales sont utilisées pour l'obtention des spectres infrarouge. La première, et la plus ancienne, est dite à onde continue (Continuous Wave ou CW) et est relativement simple à mettre en oeuvre et à comprendre, la seconde est dite à transformée de Fourier (Fourier's Transform ou FT), beaucoup plus précise, rapide, mais dont la théorie sous-jacente est moins accessible.
Dans un premier temps, nous allons voir comment se prépare un échantillon pour le spectre infrarouge, puis, nous étudierons les caractéristiques communes aux deux types de spectromètres, avant de traiter leurs aspects spécifiques.
Préparation d'un échantillon Infrarouge
Suivant la nature de l'échantillon, solide ou liquide, les techniques diffèrent. Un solide sera broyé en présence de bromure de potassium (qui est transparent jusqu'à 400 cm-1) puis comprimé sous pression réduite pour former une fine pastille. Une autre technique consiste à disperser le solide dans une paraffine (le nujol) et à déposer la suspension sur une pastille de chlorure de sodium monocristallin (transparent jusqu'à 625 cm-1).
Un liquide sera déposé entre deux pastilles de chlorure de sodium monocristallin comprimées, de manière à obtenir un film fin, ou placé dans une cuve dont les fenêtres seront des monocristaux de chlorure de sodium ou de fluorure de calcium (qui a l'avantage de ne pas être altéré par l'eau). Dans le cas des liquides purs, l'épaisseur de la cuve est souvent trop importante pour obtenir un spectre de qualité satisfaisante.

Une autre technique, valable aussi bien pour les liquides que les solides, consiste à préparer une solution diluée du produit dans un solvant, puis à étudier cette solution dans les cuves précédemment décrites. Il faut noter que tous les solvants possèdent des bandes d'absorption en infrarouge et qu'il est nécessaire de compenser ses bandes par une référence. La compensation n'étant pas toujours parfaite, les solvants utilisés pour les solutions sont choisis pour ne pas présenter de bandes d'absorption dans les zones particulièrement intéressante du spectre. Les plus couramment employés sont le tétrachlorure de carbone, le chloroforme et le sulfure de carbone (CS2).
Enfin, une dernière technique (ATR, Attenuated Total Reflection), qui gagne de plus en plus en popularité, consiste à utiliser les propriétés des ondes électromagnétiques à un dioptre formé d'un cristal de germanium ou de séléniure de zinc et du produit à analyser, dans les conditions de réflexion totale. Dans ces conditions, il se crée dans la partie produit du dioptre une onde évanescente sur une faible épaisseur de peau, qui est absorbée en partie par le produit. Le spectre obtenu après calcul permet de conduire au spectre classique en transmittance.

Caractéristiques communes des différents types de spectromètres
Les différences entre les divers types de spectromètres portent sur la manière dont le spectre est extrait de l'expérience proprement dite. Mais certains éléments sont nécessaires quelle que soit la technique utilisée : la source de radiation et le détecteur de signal.
En infrarouge, il est nécessaire de distinguer deux types de sources en fonction de la région spectrale étudiée : pour l'infrarouge proche, un filament céramique dopé avec des oxydes de lanthanide (filament de Nernst) dont le comportement se rapproche de celui du corps noir est utilisé, ou, plus fréquemment, d'un Globar (carbure de silicium).
La détection du signal est assurée par un composant assurant la conversion de la radiation incidente en un signal électrique, de préférence modulé pour une amplification et un traitement plus facile. En infrarouge, le détecteur utilisé dans est de type thermique. Classiquement, un thermocouple dont la jonction a été noircie était utilisé, mais aujourd'hui, le détecteur le plus utilisé est un composé d'un cristal de phosphate de triglycine, dopé à la L-alanine dont la polarisation varie en fonction de la température.
Le spectromètre à onde continue
Le spectromètre se présente la plupart du temps sous la forme d'un double faisceau. Le premier est destiné à recevoir l'échantillon à analyser et le second la référence.

Les signaux de chaque faisceau sont alors combinés de manière à faire disparaître l'influence de la référence puis un système dispersif monochromateur (prisme ou réseau) sépare les différentes longueurs d'onde présentes dont l'intensité est ensuite mesurée par le détecteur. Certains appareils sont toutefois à simple faisceau, le spectre de référence étant alors enregistré avant l'expérience puis soustrait lors du traitement.
Cette technique, relativement ancienne, nécessite un temps important puisque chaque longueur d'onde doit être traitée séparément, la résolution étant d'autant plus grande que la vitesse de balayage est lente, si l'on ajoute la perte d'énergie associée et la faible sensibilité (le rapport signal sur bruit peut être amélioré en enregistrant plusieurs fois le même spectre et en additionnant les signaux obtenus, mais le temps d'acquisition est multiplié d'autant), on peut aisément comprendre que cette technique soit aujourd'hui de plus en plus abandonnée au bénéfice des appareils à transformée de Fourier.
Le spectromètre à transformée de Fourier
Le spectromètre à transformée de Fourier est basé sur un interféromètre de Michelson. Lors de l'acquisition du spectre, un miroir mobile coulisse régulièrement conduisant à une figure d'interférence qui est enregistrée par le détecteur en fonction de la différence de marche p.
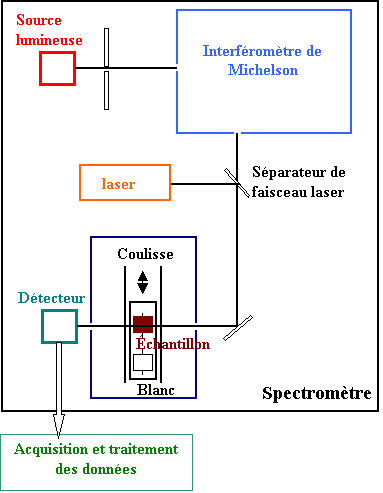

Chaque longueur d'onde interfère séparément (cohérence temporelle) et conduit à une intensité résultante :
Le signal mesuré par le détecteur correspond à la somme des intensités relatives à chaque longueur d'onde :
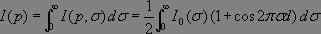
où I0(σ) représente le spectre attendu.
Posons 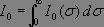 , ce qui correspond à l'intensité lumineuse mesurée à p = 0, il reste alors à calculer le terme
, ce qui correspond à l'intensité lumineuse mesurée à p = 0, il reste alors à calculer le terme 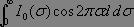 ,intégrale de Fourier où les variables d et Ï? sont conjuguées l'une de l'autre, une transformée de Fourier conduit alors immédiatement au spectre désiré :
,intégrale de Fourier où les variables d et Ï? sont conjuguées l'une de l'autre, une transformée de Fourier conduit alors immédiatement au spectre désiré :
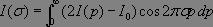
L'avantage de cette technique est que l'ensemble des longueurs d'onde est étudié simultanément, ce qui conduit à un gain de temps important et permet l'acquisition de plusieurs spectres augmentant le rapport signal/bruit de celui-ci. La résolution est aussi meilleure : il est possible de montrer que la résolution est inversement proportionnelle à la différence de chemin optique maximale entre les deux bras de l'interféromètre. Ainsi, une résolution de 0,1 cm-1 ne nécessite qu'un déplacement du miroir que de 5 cm.
- Méthodes spectroscopiques pour la chimie organique. Masson. Paris, 1997.
- Chimie Physique. DeBoeck Université. 2000.
- Spectroscopie. Dunod. Paris, 2003.
- Mécanique quantique. Tomes I et II, collection enseignement des arts, Hermann, Paris. 1998.
- Identification Spectrométrique de composés organiques. DeBoeck Université. 1998.