L’idée que l'évolution d'un système possède une direction privilégiée est familière. Le transfert de chaleur d'un corps chaud vers un corps froid, la diffusion d'une goutte de colorant dans un verre d'eau, la transformation du bois en gaz et en cendres lors de sa combustion, la corrosion de métaux au contact de l'atmosphère ou de l'eau, la décomposition d'un organisme mort : autant de phénomènes physiques, chimiques, biologiques qui appartiennent à l'expérience de tous.
Introduction
L’idée que l'évolution d'un système possède une direction privilégiée est familière. Le transfert de chaleur d'un corps chaud vers un corps froid, la diffusion d'une goutte de colorant dans un verre d'eau, la transformation du bois en gaz et en cendres lors de sa combustion, la corrosion de métaux au contact de l'atmosphère ou de l'eau, la décomposition d'un organisme mort : autant de phénomènes physiques, chimiques, biologiques qui appartiennent à l'expérience de tous.
Tout aussi familière nous apparaît la constance des êtres vivants qui ne changent que lentement du fait de leur croissance ou de leur vieillissement. Ces derniers semblent ainsi échapper aux contraintes gouvernant l'évolution des systèmes décrits au paragraphe précédent. Compte tenu d'autres singularités observées en biologie (organisation, composition, reproduction,…), on a longtemps considéré que les lois gouvernant le fonctionnement du monde inerte ne s'appliquaient pas au monde vivant. Il n'en va plus de même aujourd'hui. Le monde vivant constitue sans conteste un mode d'organisation de la matière d'une complexité inouïe ; comme on le verra dans cet article, il n'est cependant en rien singulier quant aux principes qui déterminent son fonctionnement.
Une expérience simple, aisément réalisable à son domicile, sert à illustrer le propos. Dans une première phase de l'expérience, on ajoute grain par grain du gros sel de cuisine dans un grand verre contenant V = 10 cl d'eau. On en observe le contenu après agitation destinée à faciliter la dissolution. Au début, le sel se dissout aisément. Après addition d'une masse d'environ 4 g[1], il demeure un peu de sel non dissous au fond du verre (figure 1). On note alors le nombre total n1 de grains de sel ajoutés depuis le début de l'expérience. On ajoute dans le verre un volume V = 10 cl d'eau, ce qui provoque la dissolution du solide résiduel, et on reprend l'addition des grains de gros sel de sorte à mesurer le nombre total de grains n2 qu'il faut introduire pour provoquer la réapparition de solide résiduel au fond du verre. On achève cette première phase de l'expérience en notant avec un feutre le niveau de l'eau dans le verre.

Figure 1. a) Gros sel de cuisine (photographie : J. Dodémont). b) Lorsque l'on introduit 4 g de sel dans 10 cL d'eau, on observe au fond du verre quelques cristaux de sel non dissous (photographie : S. Thlang).
Dans une seconde phase de l'expérience, on ajoute suffisamment d'eau dans le verre de façon à dissoudre le sel solide résiduel (5 à 10 cl). On abandonne ensuite ce verre dans un coin de la cuisine durant une dizaine de jours. Au bout d'un certain temps, on observe que des petits cristaux incolores se sont formés[2]. On note alors au feutre le niveau de l'eau.
Au début de la première phase de l'expérience, le sel ajouté se dissout intégralement dans le volume d'eau V choisi (il ne disparaît pas ; voir ci-dessous). Au-delà d'une certaine quantité de sel ajoutée, n1, l'eau ne peut dissoudre le trop plein de sel : la solution est saturée. On conclut ainsi que la capacité de l'eau à dissoudre le sel est limitée. Si on a manipulé proprement et si les grains de sels sont tous de calibres voisins, on doit par ailleurs observer que n1= n2. Ceci suggère que la concentration en sel, c'est-à-dire la quantité de sel apportée divisée par le volume d'eau, est la grandeur permettant de caractériser la capacité de l'eau à dissoudre le sel. Comme :
n1/V = (n1+ n2)/2V
on remarque en effet que la saturation de la solution se manifeste quand la concentration est égale à n1/V. La coexistence du sel solide et de la solution aqueuse semble ainsi associée à une concentration caractéristique qui ne dépend ni du volume d'eau V, ni de la quantité de sel apportée mais d'un rapport de ces deux paramètres.
Dans la seconde phase de l'expérience, rien de notable se produit initialement lorsqu'on laisse s'évaporer la solution contenant le sel intégralement dissous. Cependant, lorsque la quantité d'eau résiduelle devient égale à la quantité d'eau 2V utilisée dans la première phase de l'expérience (comme on le vérifie en notant que les traits de feutres tracés lors des deux phases de l'expérience sont sensiblement situés au même niveau), il se forme des cristaux de sel. Dans cet état, on remarque que la concentration (n1+ n2)/2V est égale au rapport caractéristique mis précédemment en évidence. Il se confirme ainsi que la coexistence du sel solide et de la solution saline est associée à une concentration singulière de sel dans l'eau, appelée solubilité en chimie.
L'évolution d'un mélange initialement constitué de sel solide et d'eau dépend ainsi des quantités relatives de sel et d'eau mis en présence. Le sel placé dans l'eau se dissout intégralement tant que sa concentration finale demeure inférieure à sa solubilité. Au-delà, la solution est saturée : solide et solution coexistent. L'augmentation de la concentration de sel en solution qui accompagne l'évolution de tout mélange de sel solide et d'eau tend donc à le rapprocher d'un état « d'équilibre » caractérisé par une concentration : la solubilité du sel dans l'eau.
À l'issue de l'expérience précédente, le solide en excès au fond du verre semble passif ; apparemment plus rien ne se passe. Une approche plus fine, à l'échelle des atomes ou molécules, dépassant les limites de notre perception montre qu'il n'en est rien.
Le sel de cuisine est principalement constitué de chlorure de sodium solide qui contient deux ions, sodium Na+, et chlorure Cl-, empilés de manière périodique (voir Figure 2). L'eau est quant à elle un liquide constitué de petites molécules contenant deux atomes d'hydrogène liés chacun à un atome d'oxygène (Figure 3)[3].

Figure 2. Gauche : cristal de hyalite (chlorure de sodium natif) ; droite : à l'échelle atomique, le sel de cuisine contient principalement du chlorure de sodium résultant d'un empilement compact d'ions sodium (positivement chargé ; en jaune) et chlorure (négativement chargé ; en vert).
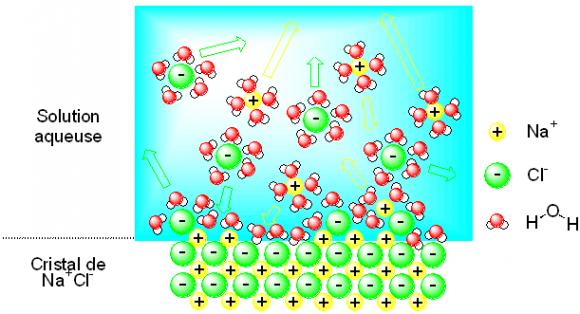
Figure 3. Description de la dissolution et de la précipitation du chlorure de sodium dans l'eau. Certains ions, animés de mouvements permanents, sont pris en charge par des molécules d'eau et passent en solution ; ce processus tend à dissoudre le cristal. Dans le même temps, certains ions dans un nid de molécules d'eau chutent sur la surface des cristaux et s'y fixent après avoir perdu les molécules d'eau qui les enrobent ; ce processus tend à accroître le cristal. L'évolution de la position de la frontière entre le cristal de chlorure de sodium et la solution aqueuse dépend des vitesses de dissolution et de précipitation. Les molécules d'eau ne sont pas représentées intégralement en solution aqueuse afin de faciliter la lisibilité de la figure. La solution aqueuse représentée ici symboliquement par le rectangle bleu est en réalité densément peuplée de molécules d'eau.
Le monde atomique n'est pas immobile, loin s'en faut. À température ambiante, atomes, ions et molécules sont animés de mouvements désordonnés incessants : les ions sodium et chlorure oscillent faiblement autour de leur position moyenne dans le cristal, les atomes d'hydrogène et d'oxygène de la molécule d'eau explorent des géométries légèrement différentes du coude parfait représenté à la Figure 3, les molécules d'eau se déplacent continûment les unes par rapport aux autres,... Lorsqu'on place un cristal de chlorure de sodium dans de l'eau, ions et molécules d'eau se comportent comme les auto-tamponneuses à la fête foraine : l'interface entre le cristal et l'eau est l'objet de chocs violents. Lorsque les chocs sont appropriés, des molécules d'eau parviennent à arracher des ions de la surface du cristal (Figure 3). Le nombre de paires d'ions extraites ainsi du cristal par unité de surface et par unité de temps, v-, dépend en fait essentiellement de la température. Ce nombre gouverne la vitesse de dissolution du cristal. Certains ions sodium et chlorure contenus dans la solution chutent quant à eux tels des météorites sur la surface du cristal ; le plus souvent, ils rebondissent mais parfois, ils restent piégés par la surface et viennent s'insérer dans le cristal (Figure 3). Le nombre de paires d'ion sodium et chlorure incorporés par unité de surface et par unité de temps dans le cristal, v+, dépend lui aussi de la température, mais il est en outre proportionnel aux concentrations en ions sodium et chlorure contenus dans la solution. C'est ce nombre qui contrôle la vitesse de précipitation du cristal.
Lors de la première phase de l'expérience, on ajoute un cristal de sodium dans un volume donné d'eau pure ; on a dissolution du cristal à la vitesse v-. À ce même instant, la vitesse de précipitation v+ est par ailleurs nulle puisqu'il n'y a initialement pas d'ions sodium et chlorure présents dans la solution. Au bilan qui fait intervenir la différence des vitesses de dissolution et de précipitation, v- - v+, le cristal disparaît puisque la vitesse de dissolution est supérieure à la vitesse de précipitation. Au fur et à mesure des additions successives de sel, le solide apporté continue à disparaître et la solution s'enrichit progressivement en ions sodium et chlorure. L'augmentation correspondante des concentrations des deux ions accroît ainsi v+ sans modifier v-. Pour des valeurs suffisamment grandes des concentrations en ions sodium et chlorure, les vitesses de dissolution et de précipitation deviennent égales : v+ = v-. Dans cet état, tout nouveau cristal apporté est stable, c'est-à-dire qu'il ne se dissout pas puisque sa vitesse de dissolution est égale à sa vitesse de précipitation : l'état d'équilibre est atteint vis-à-vis du processus de dissolution. Comme l'illustre la Figure 3, cet état d'équilibre ne doit cependant pas s'interpréter comme un état de mort ; la surface du cristal est l'objet d'échanges de matière incessants. Les vitesses de dissolution et de précipitation demeurent toutes deux non nulles dans l'état d'équilibre que l'on qualifie de dynamique.
On s'intéresse maintenant à la seconde phase de l'expérience. Le volume considéré de solution salée sous-saturée, c'est-à-dire qui ne contient pas de solide, contient désormais des quantités d'ions sodium et chlorure telles que, si un petit cristal venait à se former, on aurait une vitesse de dissolution supérieure à la vitesse de précipitation: v- > v+. Dans de telles conditions, tout cristal est instable de sorte que l'on n'observe initialement pas l'apparition de solide. Au cours de l'évaporation de la solution, la concentration en ions sodium et chlorure augmente puisque la quantité d'ions contenue dans le verre est fixe alors que le volume de la solution diminue ; v+ s'accroît au cours de l'évaporation. Lorsque les concentrations en ions deviennent suffisamment élevées, les vitesses de dissolution et de précipitation deviennent égales : v+ = v-. Lorsque cet état est atteint, la formation d'un petit cristal ne s'accompagne plus de sa dissolution. Toute évaporation supplémentaire se traduit par la croissance du cristal ou par la formation d'un nombre plus grand de cristaux de sorte que l'égalité v+ = v- est maintenue quelle que soit la quantité de solide formé.
L'analyse à l'échelle des atomes, des ions et des molécules permet ainsi de préciser l'interprétation de l'état d'équilibre vis-à-vis du processus de dissolution introduit précédemment : l'état d'équilibre se caractérise par l'égalité des vitesses des processus inverses de dissolution et de précipitation.
Les paragraphes précédents dégagent les caractéristiques de l'évolution de nombreux systèmes en chimie :
-
un système évolue sous l'action des réactions chimiques dont il est l'objet (dans l'exemple choisi, la réaction de dissolution et la réaction de précipitation). Ces réactions chimiques vont toujours par paire : une réaction directe qui mène des réactifs aux produits (par exemple, la réaction de dissolution qui convertit le chlorure de sodium solide en ions), et une réaction inverse qui mène des produits aux réactifs (dans ce cas, la réaction de précipitation qui convertit les ions en chlorure de sodium solide);
-
l'issue de cette évolution est un état d'équilibre. Cet état est caractérisé par un jeu de valeurs des concentrations des différents réactifs et produits présents (ici, la solubilité du chlorure de sodium dans l'eau). Dans l'état d'équilibre, les vitesses des réactions directe et inverse au sein d'une paire, non nulles, sont égales.
Les paragraphes précédents ne contiennent pour autant aucune clef permettant de dégager le principe responsable de l'évolution. Deux considérations sont impliquées lorsqu'il s'agit d'aborder cet aspect.
-
On remarque tout d'abord sur la Figure 3 que la dissolution des ions sodium et chlorure s'accompagne de changements d'environnement pour ces ions eux-mêmes, mais aussi pour des molécules d'eau. Dans un mélange de sel solide non dissous et d'eau, les ions sodium et chlorure sont environnés par d'autres ions sodium et chlorure, et les molécules d'eau sont quant à elles environnées de molécules d'eau[4]. Dans une solution de chlorure de sodium, les ions sont entourés par des molécules d'eau et les molécules d'eau sont parfois au contact d'ions. Ces changements d'environnement modifient la répartition de l'énergie du système qui dépend précisément des positions relatives des atomes les uns par rapport aux autres.
-
La seule information relative aux redistributions d'énergie lors du processus de dissolution/précipitation ne permet pas de déterminer la direction dans laquelle s'effectue l'évolution du système qui y est soumis. Cette évolution implique en effet aussi la diversité des situations que recouvre les adjectifs, “solide” et “dissous”, utilisés seuls de façon dichotomique à notre échelle pour décrire l'intégralité des multiples configurations du mélange de chlorure de sodium et d'eau. Les Figures 2 et 3 montrent que les ions sodium et chlorure sont distribués de façon unique dans l'état “solide”. En revanche, il existe un nombre beaucoup plus important de configurations de ces ions associées à l'état invariablement qualifié de “dissous” ; ces configurations se distinguent, par exemple, par la position des ions en solution.
L'évolution spontanée d'un système s'effectue toujours dans une direction qui rend son état de désordre maximum : un état est ainsi toujours plus ordonné que celui qui le suit au cours de son évolution, et l'état d'équilibre est le plus désordonné des états accessibles au système. Dans un système qui peut facilement échanger de l'énergie avec son environnement, les états atteints avant l'équilibre peuvent faire intervenir une proportion trop grande de configurations de haute énergie, ou bien un nombre insuffisant de configurations, ou bien encore une combinaison de ces deux causes. Dans une solution sous-saturée, la formation d'un cristal de chlorure de sodium s'accompagnerait ainsi d'une plus grande singularité de l'organisation des atomes contenus dans le verre, tout comme la dissolution d'un cristal de chlorure de sodium dans une solution sur-saturée.
Une analogie exploitant le sens commun est peut-être ici utile. Considérons une étagère dans une bibliothèque. Sur cette étagère, toutes les configurations associées aux différents arrangements de livres sont d'énergie identique. L'état « rangé » correspond à une configuration unique, issue d'un classement par ordre alphabétique par exemple. Toute autre configuration (et il en existe un nombre important !) appartient à l'état que nous qualifions de « désordonné », sans spécification supplémentaire. Un(e) bibliothécaire range initialement l'étagère. On s'intéresse par la suite à son évolution spontanée, c'est-à-dire sans intervention régulatrice, une fois soumise aux emprunts/retours de visiteurs peu consciencieux (du genre molécule agitée !). Elle atteint souvent assez vite son état d'équilibre vis-à-vis de ce processus de redistribution et il s'agit de l'état désordonné ! Il est de même énergie que l'état rangé initial, mais il est associé à un plus grand nombre de configurations et il se révèle donc plus probable. L'état « livres de l'étagère répandus sur le sol de la bibliothèque » constituerait un état encore plus probable du système car d'énergie plus faible que l'état étagère désordonnée précédent. Il devrait donc constituer l'état d'équilibre du système. Il faudrait cependant pour cela l'intervention d'un nouveau processus de redistribution dans lequel les livres ne sont plus seulement échangés au sein de l'étagère, mais entre le sol et l'étagère. On suppose ici que les utilisateurs de la bibliothèque sont peu consciencieux mais néanmoins civilisés et on a donc exclu l'existence de ce nouveau processus. Cette digression fournit l'occasion de préciser que l'état d'équilibre d'un système n'est interprétable qu'à la lumière des processus gouvernant son évolution ; il faut parler d'état d'équilibre vis-à-vis d'un processus d'évolution.
Plusieurs acteurs de l'évolution des systèmes chimiques sont désormais identifiés : l'état initial qui fixe les quantités de matière des constituants du système (les réactifs et les produits), les réactions chimiques qui constituent les processus d'évolution, et le principe d'évolution qui détermine l'état d'équilibre qui représente l'état final du système lorsqu'il est atteint. Pour ce qui est de l'histoire du système, on connaît ainsi le début et la fin ; il nous manque cependant encore une clef de lecture nous permettant de disposer de l'intégralité du récit.
La section précédente a montré que tout système placé dans un état de hors équilibre est susceptible d'évoluer ; il possède en fait un “potentiel d'évolution” qui fait intervenir à la fois l'énergie et sa répartition au sein du système. Ce potentiel peut être illustré grâce à une fructueuse analogie hydrostatique (Figure 4). Considérons à nouveau l'opération de dissolution d'un cristal de chlorure de sodium telle qu'elle est réalisée dans la première phase de l'expérience ci-dessus. L'état initial du système est constitué d'un cristal de chlorure de sodium et d'un certain volume d'eau pure. Cet état hors équilibre est assimilé à l'état d'un barrage séparant deux compartiments contenant de l'eau à des niveaux différents (Figure 4a). Les compartiments de gauche et de droite représentent respectivement les réactifs (ici le chlorure de sodium solide) et les produits (ici le chlorure de sodium dissous) de la réaction chimique étudiée (ici la réaction de dissolution). Au cours de son évolution, du chlorure de sodium solide donne du chlorure de sodium dissous sous l'action de la réaction de dissolution ; le système évolue vers son état d'équilibre. De même, l'eau s'écoule du compartiment de niveau le plus haut vers le compartiment de niveau le plus bas lorsqu'on ouvre une vanne séparant les deux compartiments du barrage ; cet écoulement cesse lorsque les niveaux de part et d'autre du mur sont identiques : l'équilibre est atteint vis-à-vis de l'écoulement de l'eau au travers de la vanne.
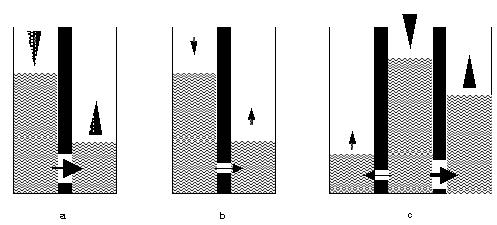
Figure 4. a). La transformation associée à l'écoulement de l'eau s'effectue spontanément de gauche à droite et à vitesse élevée en raison du grand diamètre de la vanne. L'écoulement s'arrête lorsque les niveaux d'eau de part et d'autre du barrage sont identiques ; b). même légende que a mais avec un écoulement plus lent ce qui augmente la durée de l'évolution sans toutefois modifier l'état d'équilibre atteint; c). L'eau du compartiment central s'écoule vers les compartiments latéraux. Dans le cas considéré, l'eau s'écoule rapidement vers le compartiment de droite et plus lentement vers le compartiment de gauche. Dans un premier temps, on va assister à l'égalisation des niveaux des compartiments central et latéral droit. Dans un second temps, il y aura égalisation du niveau d'eau dans les trois compartiments.
On imagine facilement que si l'on réduit le diamètre de la vanne, l'eau s'écoule toujours de gauche à droite mais plus lentement ; un même état d'équilibre sera cependant atteint (Figure 4b). De même, la vitesse d'une réaction chimique modifie la durée de l'évolution du système mais elle n'affecte pas la nature de l'état d'équilibre.
On considère maintenant la situation plus compliquée de la Figure 4c. Il existe désormais trois compartiments et deux barrages. Le compartiment central, analogue à des réactifs chimiques, peut se transvaser dans deux compartiments situés respectivement à sa gauche et à sa droite. Du point de vue de l'analogie, ces deux compartiments représentent les produits distincts de deux réactions chimiques dont les réactifs sont l'objet. L'état initial est défini par les quantités d'eau placées dans chacun des compartiments avant ouverture des vannes. L'état d'équilibre est celui dans lequel les niveaux des trois compartiments sont identiques. De façon intuitive, on se doute que ce sont les vitesses d'écoulement dans les deux vannes de séparation qui vont gouverner les états parcourus par les trois compartiments au cours de l'évolution. Dans le cas représenté à la Figure 4c, la vitesse d'écoulement est plus grande du compartiment central vers le compartiment latéral droit, que du compartiment central vers le compartiment latéral gauche. On égalise ainsi dans un premier temps les niveaux des compartiments central et latéral droit, puis dans un second temps les niveaux des trois compartiments. On imagine facilement que l'évolution aurait été bien différente si les diamètres des vannes avaient été changés, sans pour autant altérer ni l'état initial, ni l'état d'équilibre du système.
Nous disposons maintenant du chaînon manquant : les états parcourus par un système au cours de son évolution dépendent de la vitesse des réactions chimiques dont il est le siège. Les processus sont donc au coeur de l'évolution des systèmes chimiques ; ce sont eux qui écrivent l'histoire et c'est à cause de cela que l'auteur de cet article n'est ni une algue, ni une grenouille. Initialement, trois oeufs similaires ; trois destins cependant bien différents du fait de la cinétique chimique.
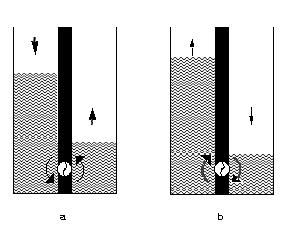
Figure 5. a). L'écoulement de l'eau s'effectue spontanément de gauche à droite ; lors de l'écoulement, il est possible de récupérer de l'énergie électrique par la mise en rotation de la turbine; b). l'écoulement de l'eau est forcé de droite à gauche par mise en rotation de la turbine grâce à un apport externe d'énergie.
Tout changement de processus ou bien seulement une modification de leurs vitesse doivent donc pouvoir provoquer une modification des états parcourus par le système. Reprenons l'analogie du barrage pour illustrer cet aspect. Dans un barrage, la conduite dans laquelle s'effectue la chute d'eau n'est pas une simple vanne comme représenté sur la Figure 4a. Elle contient une turbine dont la mise en rotation assure la production d'énergie électrique (Figure 5a). Ainsi alors que l'écoulement ne produisait rien d'utile dans la situation de la Figure 4a[5], l'énergie “potentielle” disponible dans l'état initial est partiellement convertie en énergie électrique mobilisable au cours de l'évolution représentée à la Figure 5a.
En chimie, l'homme est capable d'effectuer des conversions d'énergie similaires grâce à la mise en oeuvre de dispositifs technologiques. C'est par exemple ce qui se passe dans une pile dans laquelle chaque borne est reliée à un compartiment contenant des réactifs chimiques. Dans ce dispositif ingénieux, la conversion des réactifs en produits s'effectue avec production de travail électrique, alors qu'elle ne s'accompagnerait que de la seule production d'énergie thermique par simple mélange du contenu des deux compartiments de la pile : deux processus distincts, réaction s'effectuant par l'intermédiaire du circuit électrique grâce à la pile, et réaction directe lors du mélange, déterminent deux histoires différentes unies par un même début et par une même fin.
Le contrôle de l'évolution vers l'équilibre joue un rôle fondamental en biologie. Les êtres vivants sont en fait des usines chimiques assez singulières. Ils sont constitués de millions de réactifs/produits et sont le siège d'autant de réactions chimiques. Ils contiennent des dispositifs technologiques moléculaires, les enzymes, dont certains agissent comme des transducteurs d'énergie. Tout comme la turbine du barrage, ils convertissent l'énergie libérée lors de la réalisation spontanée d'une réaction chimique, en une autre forme d'énergie chimique associée à la synthèse de produits (par exemple le glucose à partir de dioxyde de carbone et d'eau). C'est ainsi que les êtres vivants utilisent leurs aliments à la fois comme source de briques de constitution, mais aussi comme source d'énergie.
Nous savons désormais presque tout de l'évolution des systèmes en chimie et il pourrait ainsi apparaître que l'état d'équilibre constitue le destin inexorable du monde moléculaire. Il n'en est en fait rien : le destin des molécules n'est pas inéluctable pour peu que l'on puisse, d'une manière ou d'une autre, contrôler la vitesse des réactions chimiques se produisant dans le système.
Revenons une dernière fois au barrage. Si l'on fournit de l'énergie aboutissant au contrôle du sens de rotation de la turbine, il devient possible de provoquer un flux d'eau du compartiment de niveau le plus bas vers celui de niveau le plus élevé qui excède le flux inverse « naturel » (Figure 5b). L'apport d'énergie externe permet ici de contrôler la direction d'écoulement de l'eau.
De même, le chimiste est souvent confronté à des situations dans lesquelles il cherche à imposer une direction qui s'oppose à la tendance d'évolution « naturelle » dégagée précédemment. Il y parvient parfois en fournissant une forme appropriée d'énergie. C'est par exemple la situation rencontrée lors de l'électrolyse grâce à l'apport d'énergie électrique. L'évolution spontanée d'un mélange de dihydrogène et de dioxygène s'effectue généralement ainsi dans la direction de la formation d'eau avec forte production d'énergie thermique[6]. L'électrolyse fournit quant à elle du dihydrogène et du dioxygène (figure 7) lors du passage de courant électrique dans l'eau. Cette production s'effectue en fait plus vite que la réaction spontanée de conversion du dihydrogène et du dioxygène en eau de sorte qu'au bilan, l'opération d'électrolyse permet de synthétiser du dihydrogène et du dioxygène à partir d'eau. Opposée à la tendance d'évolution naturelle, la conversion se manifestant lors de l'électrolyse apparaît comme forcée et les contraintes de l'équilibre sont dépassées.

Figure 6. On observe sur les électrodes en platine un dégagement gazeux de dihydrogène à droite et un dégagement gazeux de dioxygène à gauche. (photographie : J. Dodémont).
La biologie fait ici des merveilles qui peuplent les songes de nombreux chimistes. La photosynthèse qui met en jeu des milliers de molécules permet par exemple de forcer les réactions chimiques se déroulant au sein des végétaux. Elle ne mobilise pour cela que les sources de matière et d'énergie que sont les nutriments et le soleil. La respiration qui mobilise la seule énergie des nutriments agit de même. C'est la mise en place et l'intégration de tels processus d'orientation du destin de la matière organique qui ont permis au cours de l'évolution de notre planète l'émergence de modes d'organisation, les êtres vivants, dont la singularité résulte de l'état de hors équilibre vis-à-vis des réactions chimiques qu'ils abritent. Leur extraordinaire capacité à échapper aux contraintes de l'équilibre chimique est constitutive de la vie ; la perdre, c'est mourir.
Cet article doit beaucoup à de nombreux interlocuteurs ; je désire tout particulièrement remercier les professeurs Hervé Lemarchand, Bernard Roulet et Jacques Treiner de l'Université Paris VI. Cet article est initialement publié dans le numéro 335-336 de février-mars 2006 de la revue Découverte, la revue du Palais de la découverte.
Notes
[1] Pour indication, une cuillerée à soupe rase de sel représente environ 15 g.
[2] Si on laisse l'évaporation se poursuivre, il se forme une quantité toujours plus importante de cristaux dont le goût révèle qu'il s'agit de sel de cuisine. Ce principe d'évaporation a été appliqué à l'eau de mer et est mis en Å?uvre depuis longtemps dans les marais salants pour produire du sel.
[3] La taille d'une petite molécule telle que l'eau est d'environ un milliardième de mètre. À l'échelle humaine, ce nombre est plus facile à apprécier à l'aide d'une analogie : il y a autant de molécules d'eau dans une goutte, qu'il y aurait de petits pois dans une boîte de conserve (hypothétique !) de 100 km de diamètre.
[4] On néglige ici le rôle de l'interface eau-cristal qui n'implique qu'un petit nombre d'ions et de molécules d'eau dans le système.
[5] L'écoulement s'accompagne en réalité d'une élévation de la température de l'eau.
[6] C'est précisément cette conversion qui assure aujourd'hui une part de la propulsion des fusées.