Plusieurs métaux de transition jouent un rôle décisif en chimie biologique et en santé humaine. Nous nous intéressons ici au rôle joué par le fer dans le paludisme, ainsi qu’à la façon dont cette réactivité peut être utilisée pour mettre au point de nouveaux médicaments.
Le paludisme
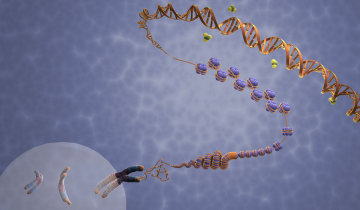
Le corps humain contient 4 à 5 grammes de fer (pour un adulte de 70 kg environ) ; 70 % de ce fer est contenu dans l’hémoglobine, protéine constituée de 4 chaînes protéiques, chacune étant accrochée à un hème (en rouge sur la Figure 1) centré sur un atome de fer (schématisé Figure 1).
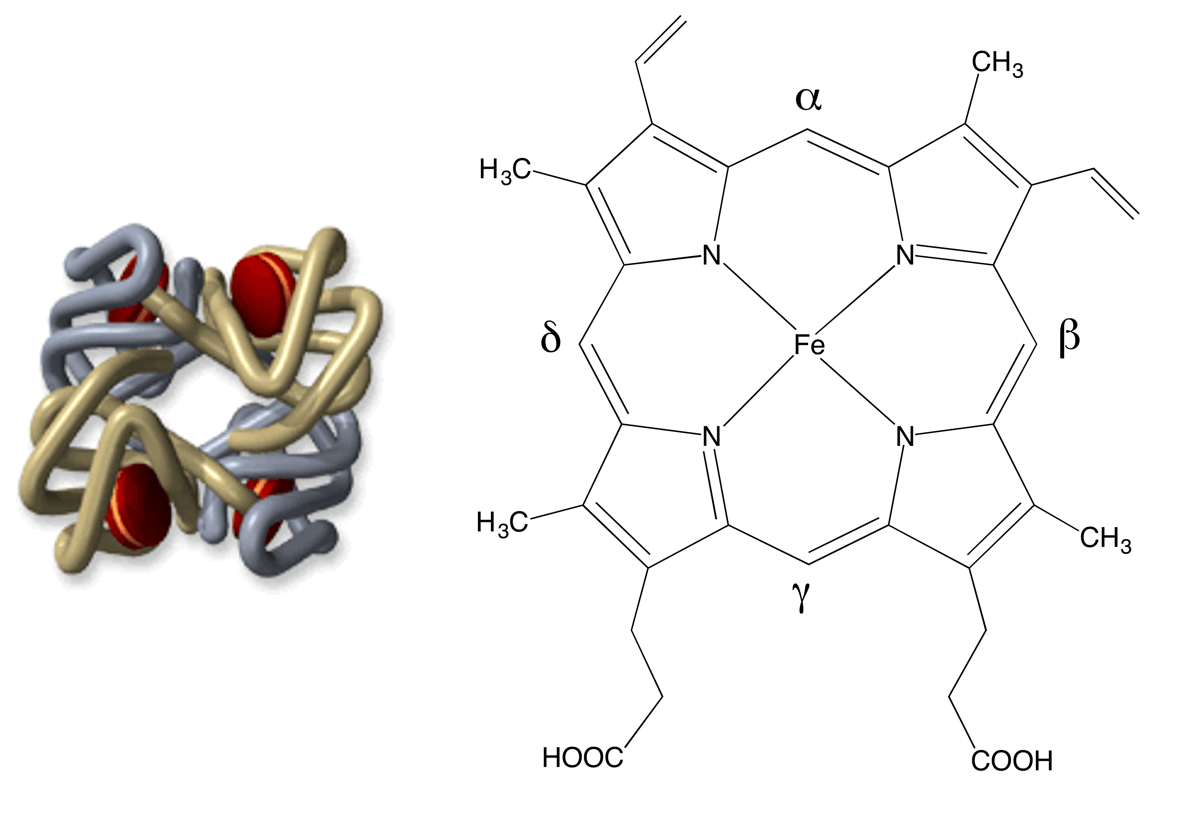
C’est la coordination du dioxygène sur le fer qui permet son transport et sa distribution dans l’organisme. Il s’agit donc d’un rôle vital. D’où l’idée, somme toute simple, de s’intéresser au rôle du fer lorsque des évènements pathologiques se produisent dans le sang.
Le paludisme (en anglais malaria) est une maladie parasitaire du sang. Cette maladie constitue un problème majeur de santé publique : 50 % de la population mondiale est exposée au risque de paludisme, dans près de 100 pays. Toute la zone tropicale est touchée. Actuellement, il provoque environ 500 000 morts par an d’après l’Organisation Mondiale de la Santé (OMS), mais ce chiffre est controversé, peut-être très sous-évalué. Il y a eu un pic de mortalité entre 1980 et 2010, dû en grande partie au développement de la résistance du parasite à la chloroquine qui était le médicament de choix.1

Le parasite responsable du paludisme est un Plasmodium transmis à l’homme par la piqûre d’un moustique Anophèle (Figure 2).
Après quelques détours, le parasite s’installe dans les globules rouges et il accomplit le cycle parasitaire responsable des symptômes du paludisme (accès fébriles intenses et rythmés). Il prolifère dans l’ensemble du système vasculaire, crée des micro-thromboses dans les capillaires de tous les organes. Dans le cerveau, ces thromboses1 peuvent se traduire par une encéphalite2 qui, dans une proportion importante de cas, conduit au coma et à la mort du patient. C’est le neuropaludisme.
Si l’on veut attaquer le parasite avec quelque chance de réussite, il faut évidemment se demander dans quel environnement il se trouve, et à quoi il passe son temps. Le globule rouge est un concentré d’hémoglobine (la concentration en hémoglobine y est de l’ordre de 2 mmol.L-1 donc celle du fer est comprise entre 8 et 10 mmol.L-1). Plasmodium a toute la machinerie enzymatique nécessaire pour découper l’hémoglobine de son hôte en acides aminés avec lesquels il construit ses propres protéines (Figure 3 à gauche). C’est un véritable parasite : il vit aux dépends de son hôte…

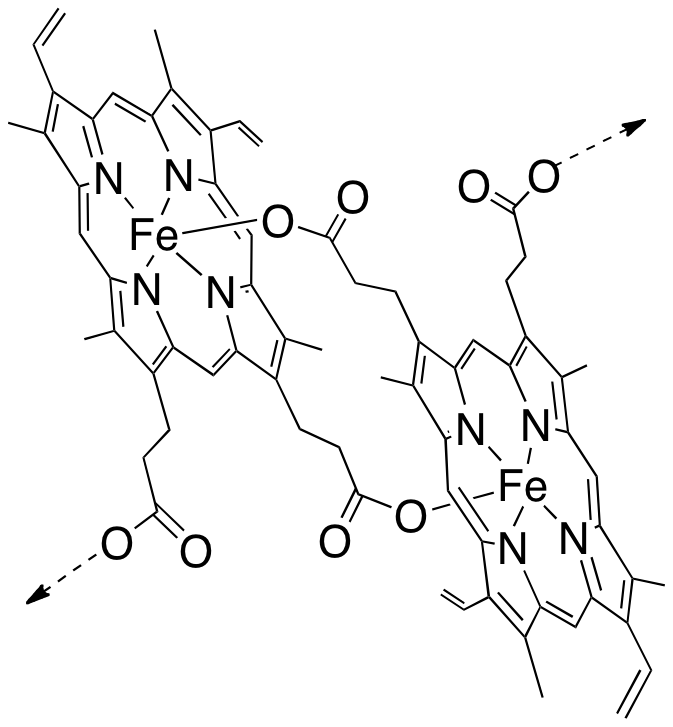
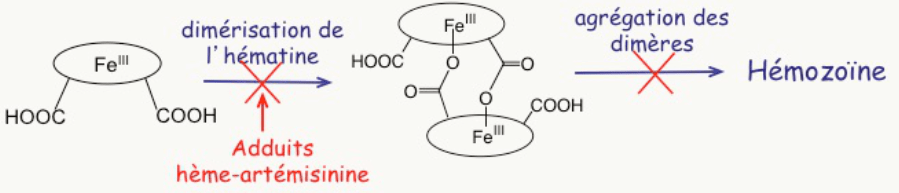
Au cours de ce processus métabolique, il libère de l’hème. Cet hème –avec son fer– était au cœur de l’hémoglobine pour transporter l’oxygène, pas pour le transformer ! Une fois sorti de la protéine, l’hème est dévoyé : son fer réagit, il réduit le dioxygène en un processus toxique pour toute cellule vivante que l’on nomme stress oxydant. Le Plasmodium doit alors échapper à la toxicité d’un déchet qu’il a lui-même généré (Figure 3 à droite). Pour cela, il polymérise l’hème-fer(II) en un pigment noir, insoluble, qui n’est plus capable de réduire l’oxygène, et n’est donc plus toxique : c’est l’hémozoïne (Figure 4).
L’hème libre –hors de la protéine–, ainsi que sa polymérisation, sont tout-à-fait spécifiques du parasite, c’est-à-dire qu’ils n’existent pas dans le globule rouge sain. L’hème et son agrégation sont donc des cibles potentielles de thérapie.
Les médicaments antipaludiques
Les dérivés synthétiques de la quinine
Les anciens médicaments antipaludiques, en particulier la chloroquine et la méfloquine (Figure 5), sont inspirés de la quinine, elle-même extraite de l’écorce de quinquina en 1820 par deux pharmaciens français, Joseph PELLETIER et Jospeh CAVENTOU. Ces composés contiennent un noyau quinoléine, plan, qui, en s’empilant avec l’hème-fer(II), empêche sa polymérisation en hémozoïne. On inhibe ainsi le système de détoxification du parasite.
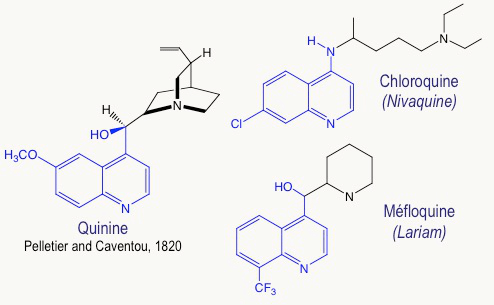
Malheureusement, les résistances à la chloroquine, et également à la méfloquine, se sont généralisées, de telle sorte que ces produits conduisent à un taux inacceptable d’échecs thérapeutiques.
L’artémisinine et son mécanisme d’action
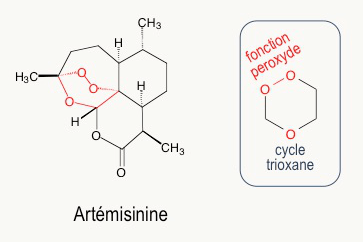
Au début des années 1970, l’artémisinine (Figure 7) est extraite des feuilles d’une armoise commune, plante utilisée depuis des siècles dans la médecine traditionnelle chinoise pour lutter contre la fièvre et, en particulier, contre le paludisme. Ces travaux ont été récompensés par le prix Nobel de médecine, en octobre 2015. L’artémisinine a une structure très différente des quinoléines. L’activité biologique est due à la fonction peroxyde d’un cycle 1,2,4-trioxane (en rouge Figure 7).1
Dans les grandes lignes, quel est le mode d’action de l’artémisinine ?
Réaction de l’artémisinine avec l’hème-fer(II)
Dans un milieu cellulaire réducteur, ce qui est le cas des globules rouges, on peut facilement envisager une réduction monoélectronique du peroxyde de l’artémisinine par l’hème-fer(II). Cette réaction (Figure 7) conduit à une homolyse du peroxyde, suivie d’une isomérisation rapide du radical alkoxy O• en radical alkyl C•. Ce dernier est un alkylant puissant qui ne va pas voyager très loin… il préfère donc, par une réaction intramoléculaire, l’hème qui lui a donné naissance.
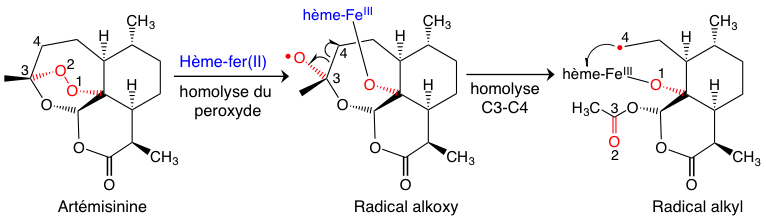
Le résultat de cette réaction très efficace est la formation de produits de couplage covalent entre l’hème et le radical alkyle issu de l’artémisinine (adduits Figure 8) [1].1
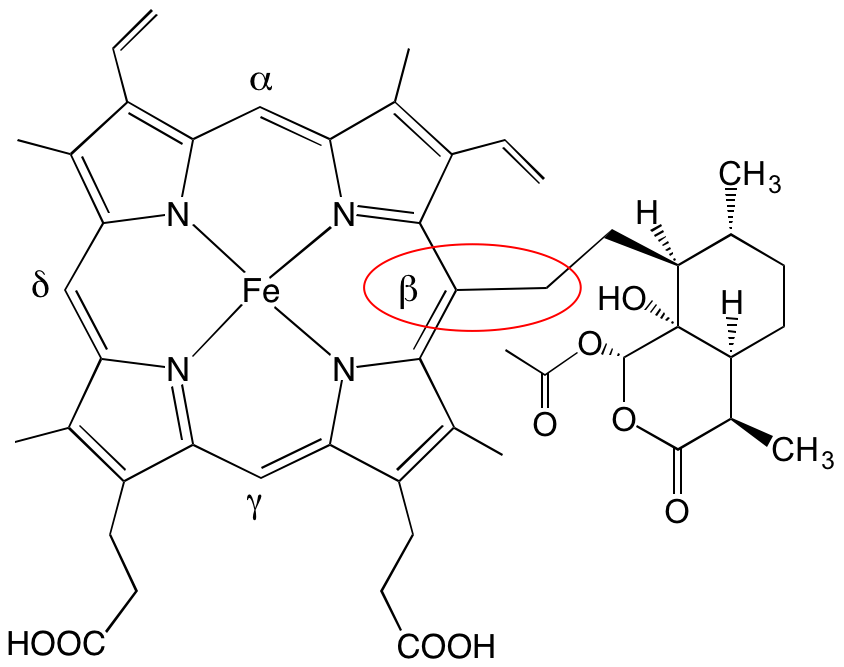
Cela se produit non seulement sur la paillasse du chimiste, mais aussi chez les souris impaludées, suggérant qu’in vivo l’hème est à la fois l’activateur et la cible du médicament [2].1 Il est important de noter que ces produits de couplage hème-médicament sont absents chez les souris saines que l’on traite dans les mêmes conditions. Cela signifie que leur présence est due à la fois au parasite et au traitement ; donc, très probablement, au mode d’action du médicament.
Comment ces adduits tuent-ils le parasite ?
Au laboratoire, nous avons vérifié que :
- ces adduits sont incapables de polymériser comme l’hème-fer(II) polymérise in vitro en b-hématine, qui est un analogue synthétique de l’hémozoïne ;
- une petite quantité de ces adduits inhibe la polymérisation de l’hème lui-même [3]. La raison en est simplement que le fragment artémisinine est volumineux et globulaire ; il constitue donc une gêne stérique (Figure 9) à l’empilement des cycles porphyriniques qui est nécessaire à la polymérisation.
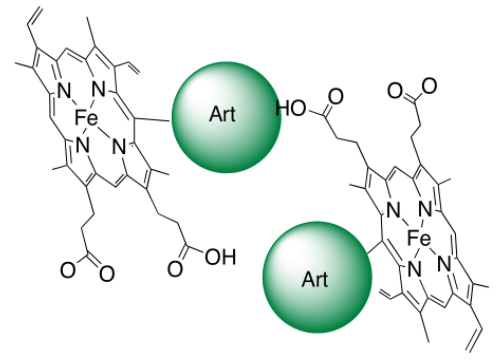
La conséquence est la suivante : dans le parasite, lorsque l’artémisinine a rencontré l’hème, il reste soit de l’hème, soit un hème modifié, tous deux solubles, non polymérisables, et dans tous les cas avec leur fer doué d’activité rédox (Figure 10). Ils réduisent l’oxygène moléculaire en superoxyde, radical anion O2–• qui constitue la première étape d’un stress oxydant fatal au parasite. 1
L’artémisinine est très efficace, y compris sur les parasites résistants à la chloroquine ou à la méfloquine. Elle est utilisée en association avec un autre antipaludique, et a sauvé des millions de vies. Malheureusement, depuis 2007-2008, des parasites répondant mal à l’artémisinine sont apparus en divers lieux de l’Asie du Sud-Est… La lutte anti-infectieuse demande des adaptations et des inventions constantes. De plus, l’armoise ne produit de bons rendements en artémisinine que lorsqu’elle pousse sur les hauts-plateaux de la Chine et du Vietnam… Problème géopolitique.
Comment imaginer de nouvelles molécules synthétiques actives, afin de s’affranchir des difficultés d’approvisionnement, en se basant sur le mécanisme d’action de l’artémisinine ?
Élaboration de molécules hybrides contenant deux pharmacophores
Nécessité d’une poly-chimiothérapie et conception des trioxaquines
Pour toutes les grandes endémies infectieuses, une poly-chimiothérapie (au moins une bithérapie) est préconisée, non seulement pour obtenir une guérison rapide et complète, mais aussi pour éviter ou retarder l’émergence de souches pathogènes résistantes aux médicaments utilisés.1
De plus, l’administration conjointe de deux médicaments peut être à l’origine de difficultés que nous ne détaillerons pas (pharmacocinétique, métabolisme…).
Nous avons donc imaginé et synthétisé des molécules contenant deux pharmacophores –c’est-à-dire deux entités ayant chacune leur activité antipaludique propre. C’est, en quelque sorte, une bithérapie dans une molécule unique, une sorte de « fusil à deux coups » [4].
L’un des pharmacophores est, bien sûr, le cycle trioxane de l’artémisinine, pour sa réactivité vis-à-vis du fer de l’hème, et ses propriétés alkylantes ; le second est une quinoléine, puisque nous avons vu que c’est le motif des antipaludiques copiés sur la quinine. Ces motifs ont tous les deux l’hème-fer(II) pour cible, ce qui justifie le bras de jonction covalent (Figure 11), par des mécanismes différents (empilement pour les quinoléines, alkylation pour le trioxane), ce qui doit minimiser l’émergence de résistance.

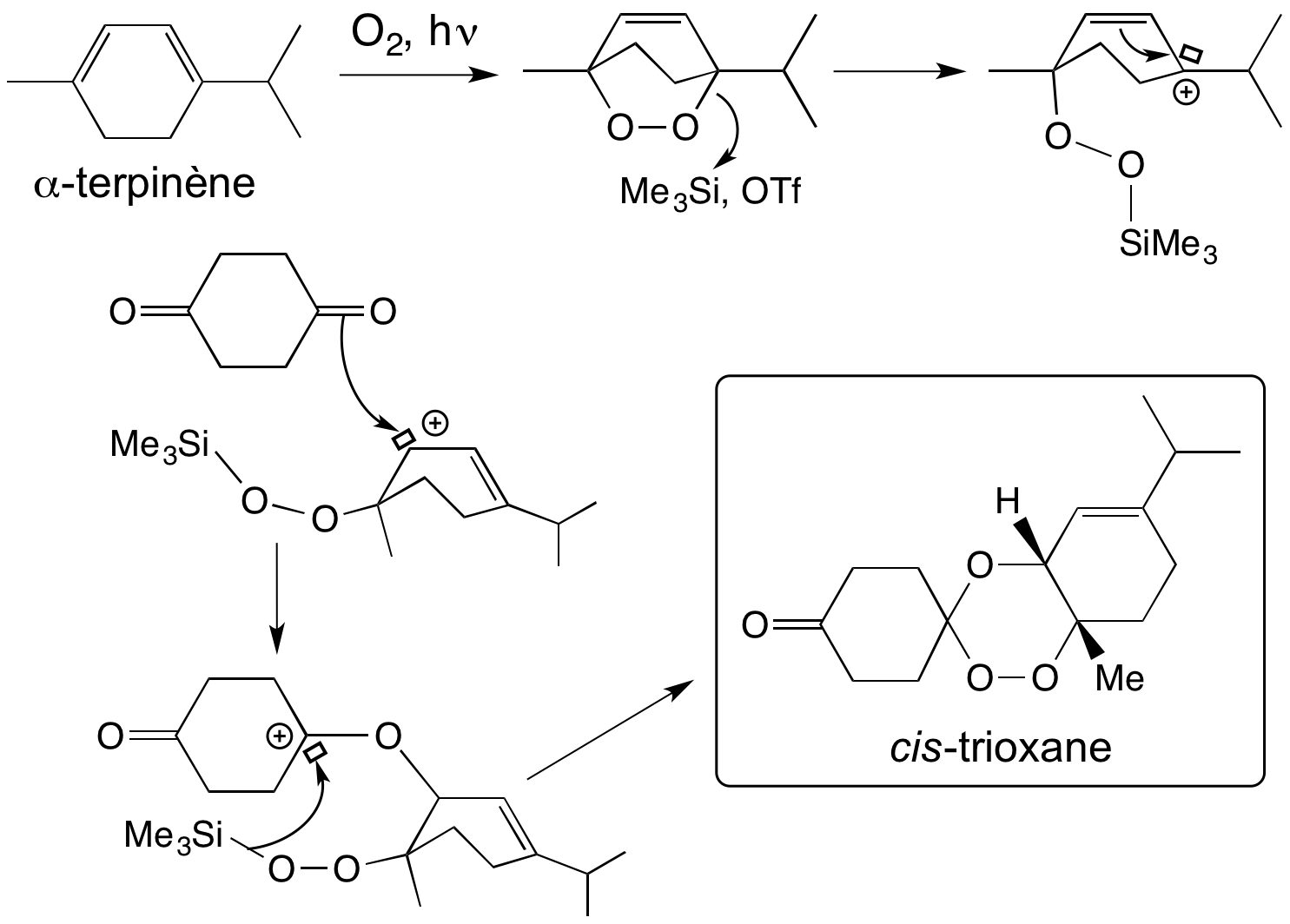
Les trioxaquines sont obtenues par une synthèse convergente simple (Figure 12), ce qui permet de multiples variations de structure. Par substitution nucléophile d’un diaminoalcane sur la 4,7-dichloroquinoléine, on obtient une 4-aminoquinoléine substituée par un bras portant une amine primaire (composé 1 de la Figure 12). Le trioxane fonctionnalisé par une cétone (composé 2) est obtenu par condensation d’un 1,4-endoperoxyde et de la cyclohexane-1,4-dione. L’amination réductrice du trioxane-cétone 2 par l’amine primaire de l’aminoquinoléine 1 conduit à la trioxaquine avec un bon rendement. Divers sels peuvent être préparés [5a-c]. (Le mécanisme de formation du trioxane est détaillé en annexe).
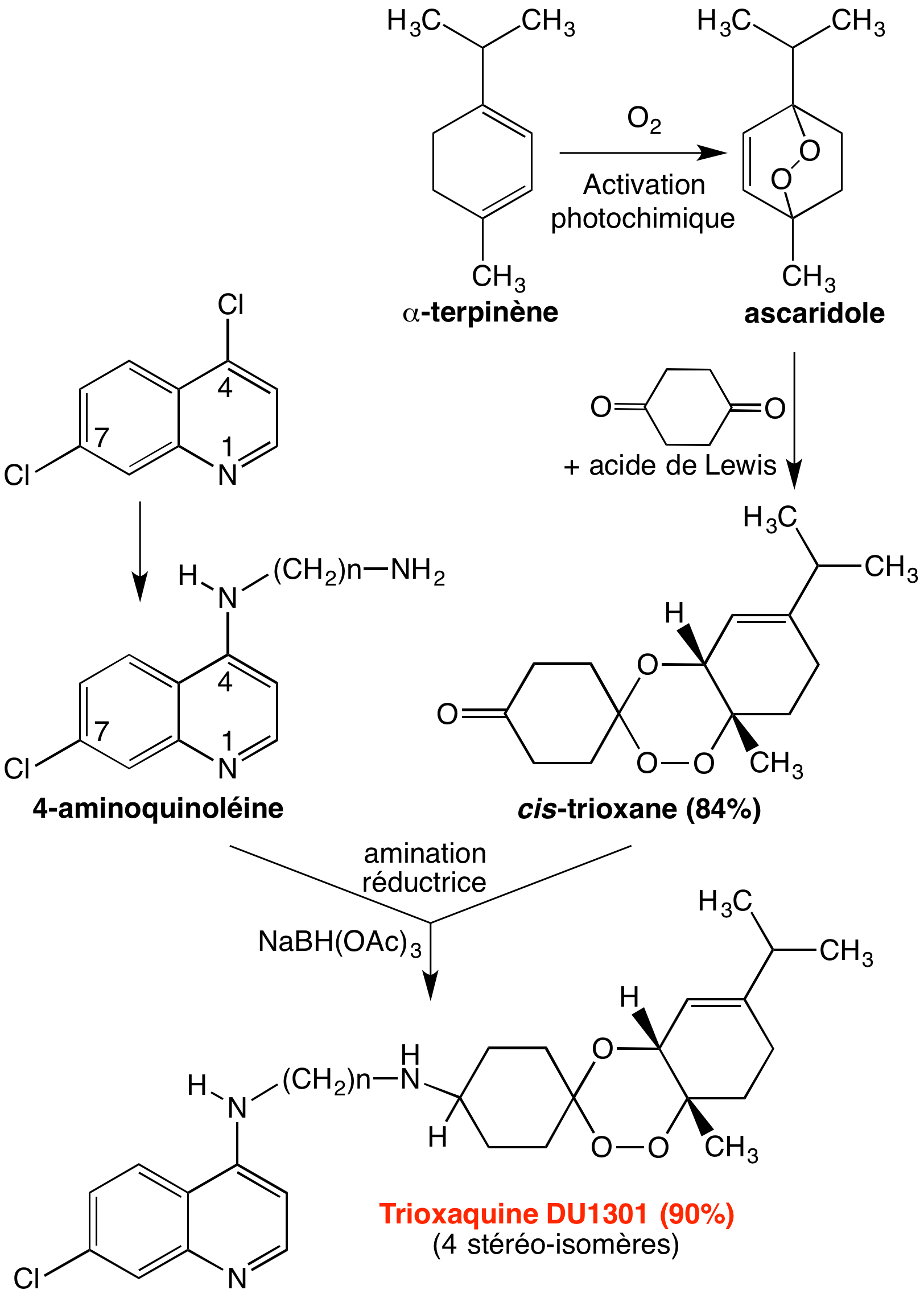
Ce schéma conduit à la synthèse d’un mélange de stéréo-isomères, du fait :
- de la chiralité de la jonction entre le trioxane et le cyclohexène d’une part ;
- de la possibilité de substitution du cyclohexane en 1,4-cis ou 1,4-trans. Chaque stéréo-isomère ayant a priori une activité biologique propre, pour éviter d’avoir à séparer, étudier et développer séparément chaque stéréo-isomère, il est souhaitable de choisir le diène et le bras de jonction de façon à limiter ou supprimer les éléments d’asymétrie de la molécule.
De plus, la synthèse doit être transposable à grande échelle et, pour un médicament visant une maladie tropicale, bon marché. C’est le cas de la trioxaquine PA1103 (ci-après) qui a été synthétisée à l’échelle de plusieurs kg, aux normes GMP (Good Manufacturing Pratices) en vigueur pour les médicaments.
Activité antipaludique des trioxaquines
Le résultat, c’est que les trioxaquines sont très actives, y compris sur les parasites qui résistent à des quinoléines synthétiques comme la chloroquine [5]. Comme l’artémisinine, elles alkylent l’hème in vitro et chez la souris impaludée [6].
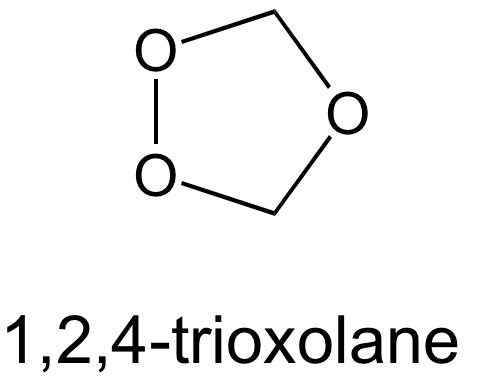
Quand on en est là, ce n’est que le tout début d’une histoire. La société Palumed a été fondée en 2000 [7]. Environ 120 trioxaquines et trioxolaquines (qui sont des analogues avec un cycle trioxolane (Figure 13) au lieu d’un trioxane) ont été synthétisées et évaluées sur des parasites en culture puis, pour les meilleures, in vivo.
De ce screening, est sortie la trioxaquine PA1103 (Figure 14) pour laquelle un accord de co-développement a été conclu avec Sanofi-aventis [7]. Cette trioxaquine est active non seulement sur les souches de laboratoire, mais aussi sur des vrais parasites de vrais malades (les « isolats cliniques »).
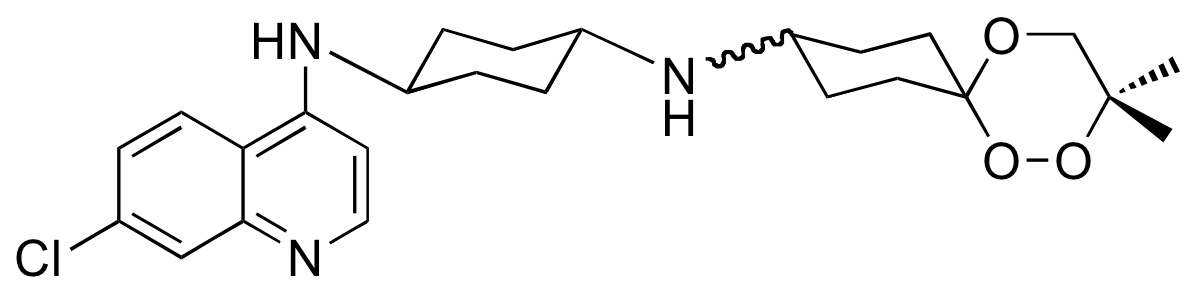
Administrée par voie orale (ce qui est important), elle est curative chez la souris, c’est-à-dire que 100 % des souris guérissent sans recrudescence de la maladie. Elle n’est ni mutagène, ni cardiotoxique. Sanofi-Aventis a mis fin au développement de la PA1103 en 2010.
Application au traitement d’autres maladies causées par des parasites hématophages
La bilharziose (ou schistosomiase)
Les raisons pour lesquelles Sanofi-Aventis a mis fin au développement d’une trioxaquine antipaludique ne sont pas scientifiques ; elles ne mettent pas en cause la pertinence de la démarche. Nous avons donc décidé d’évaluer l’activité des trioxaquines sur d’autres parasites hématophages qui ont le même processus de digestion de l’hémoglobine et de polymérisation de l’hème que Plasmodium.
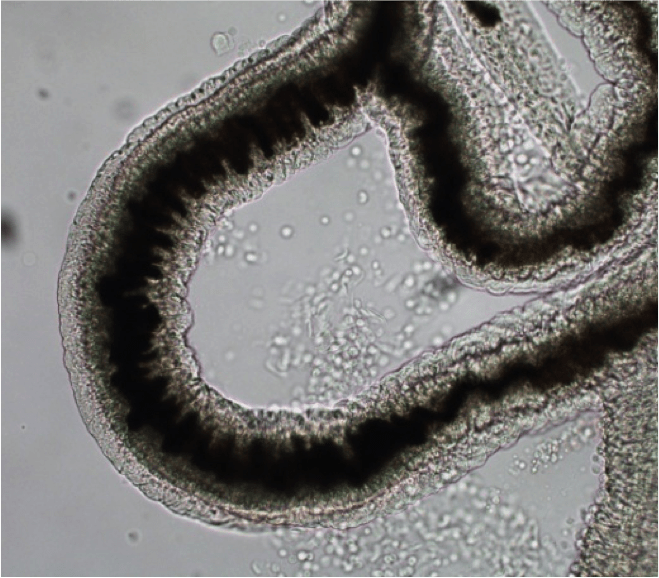
La bilharziose (ou schistosomiase) est, en nombre de cas et en nombre de morts, la deuxième parasitose tropicale après le paludisme. C’est une maladie chronique provoquée par les schistosomes, vers sexués d’environ 1 cm de long. La photo (Figure 15) représente une portion de schistosome femelle adulte ; son corps contient un long cordon noir : c’est son tube digestif, rempli d’hémozoïne.
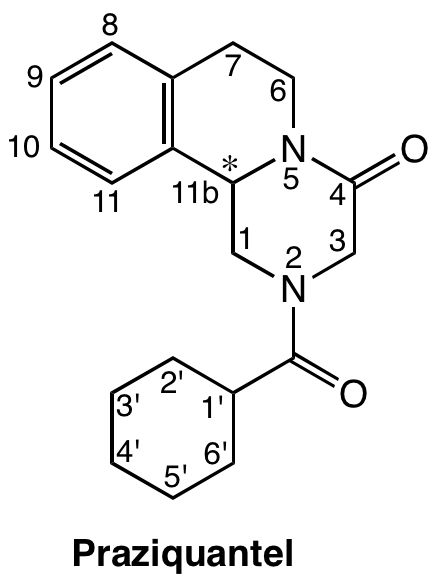
Un seul médicament est disponible, le praziquantel (Figure 16), utilisé en monothérapie depuis près de 40 ans, et pour lequel une baisse de sensibilité est observée en divers lieux [8].
Des peroxydes alkylants, comme l’artémisinine et les trioxaquines, ont-ils un effet sur ces parasites ?
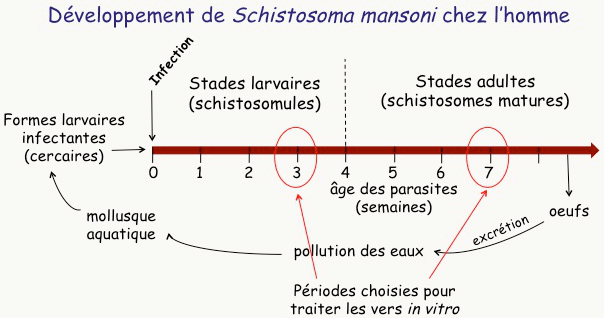
Le schistosome est un parasite beaucoup plus complexe que Plasmodium, avec un cycle de vie beaucoup plus long (9 semaines pour l’une des espèces qui infectent l’homme, S. mansoni Figure 17). Dans les eaux stagnantes de pays tropicaux, un petit mollusque émet des larves de schistosomes appelées cercaires. Lorsqu’une personne est en contact avec l’eau infestée, ces cercaires traversent la peau en quelques minutes, puis colonisent le système lymphatique et le système sanguin. Là, les larves se développent –elles prennent leur temps : 3 à 4 semaines–, deviennent des vers adultes sexués qui s’accouplent dans les vaisseaux sanguins des tractus urinaire ou digestif1. La femelle pond alors une grande quantité d’œufs qui sont responsables de la pathologie. Une petite quantité d’œufs est excrétée et, en l’absence d’installations sanitaires convenables, ces œufs vont polluer les eaux de surface et participer à la transmission de la maladie.
Dans la recherche d’une thérapie, deux paramètres doivent être considérés : d’une part, les personnes vivant en pays d’endémie sont continuellement ré-infectées, ce qui conduit les malades à être porteurs de tous les stades de schistosomes ; d’autre part, le traitement doit être le plus court possible, si possible efficace en une seule prise. Un « bon » médicament doit donc être actif sur tous les stades parasitaires. Or, les stades successifs du schistosome sont très différents d’un point de vue métabolique, ce qui complique évidemment la thérapie.
Activité des trioxaquines sur les schistosomes
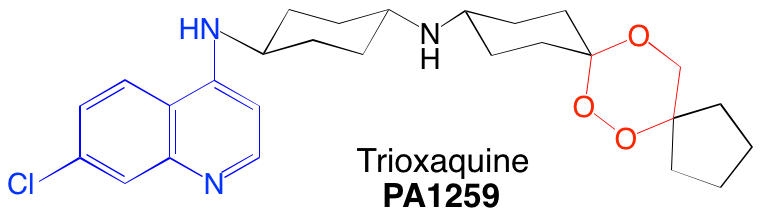
Certaines trioxaquines sont actives in vitro sur le parasite Schistosoma mansoni. Par exemple, la PA1259 (Figure 18) est active sur les stades larvaires et adultes de S. mansoni : elle contient le motif trioxane d’une part (en rouge) et le noyau quinoléine d’autre part.
Les photos suivantes (Figure 19) montre la différence entre une femelle non traitée, avec de l’hémozoïne dans son intestin aux parois bien définies et une femelle traitée par PA1259.
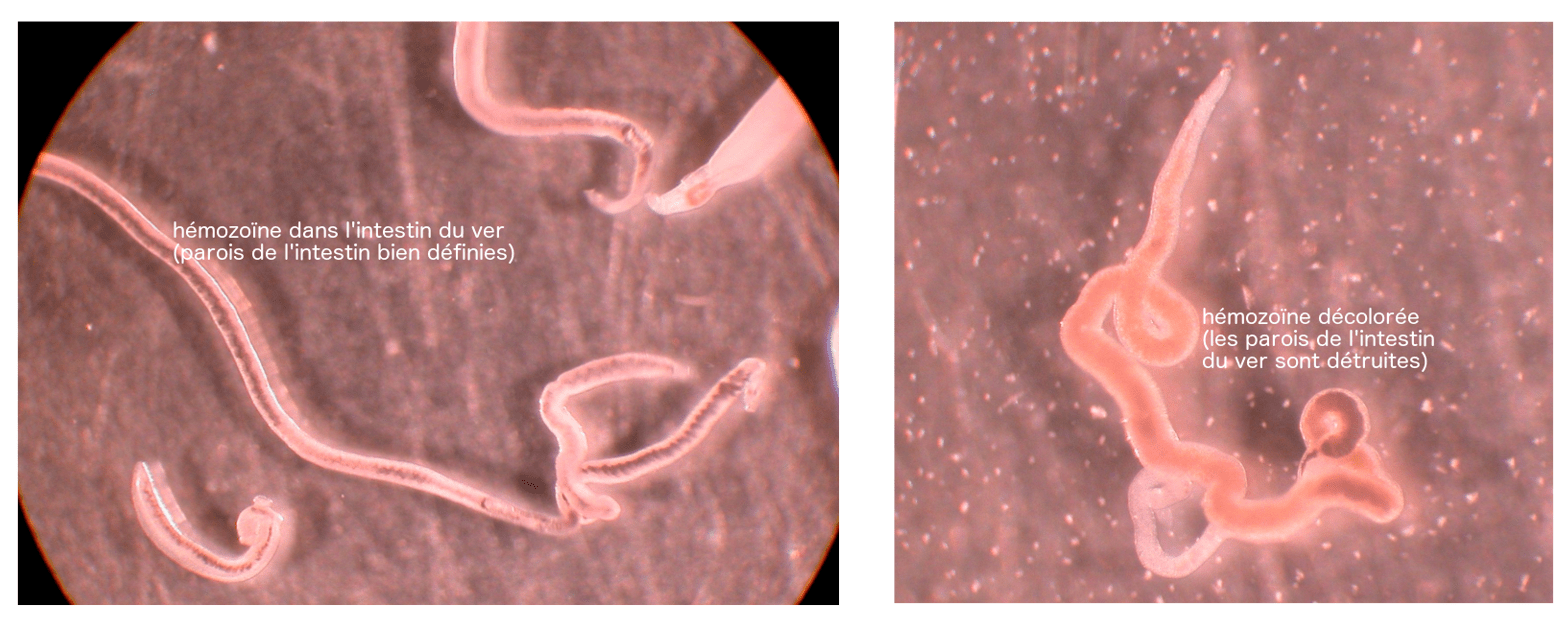
Dans ce dernier cas, non seulement le ver est mort, mais l’hémozoïne est fortement décolorée, ce qui indique la destruction de cette molécule.1 Ce qu’il reste d’hémozoïne a aussi envahi le corps du ver, preuve de dégâts occasionnés à la paroi intestinale [9]. De l’hème alkylé par la trioxaquine est également présent chez les vers traités [9]. L’hémozoïne est donc bien, dans ce cas encore, une cible du médicament comportant un peroxyde. Cette trioxaquine est active également sur les stades larvaires de schistosomes.
Vers une bithérapie trioxaquine + praziquantel
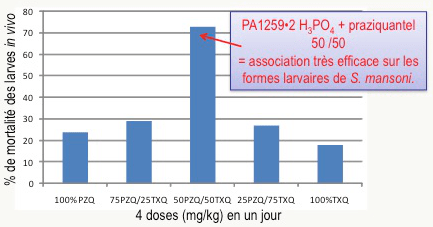
Le praziquantel, seul médicament utilisé en clinique actuellement, cible principalement les vers adultes et est peu actif sur les formes juvéniles. Par ailleurs, il son mécanisme d’action est différent de celui des peroxydes alkylants comme les trioxaquines. Pour tenter de mettre à profit une complémentarité de ces deux molécules actives, des souris malades ont été traitées par administration simultanée de trioxaquine PA1259 (sous forme de sel diphosphate) et du praziquantel. Ce traitement se traduit par un effet synergique : l’association de doses égales de ces deux médicaments est très active, y compris sur les stades larvaires, ouvrant la voie à une éventuelle bithérapie (Figure 20).
Bien que 200 millions de personnes soient infectées par la schistosomiase en divers pays (environ 200 000 morts par an), la recherche de nouveaux médicaments n’est pas considérée comme une priorité.1 Pourtant, les pays de la zone tempérée ne sont pas non plus totalement à l’abri… Le petit escargot responsable du maintien du cycle parasitaire vit à l’état endémique dans les rivières de Corse. Plus d’une centaine de personnes ayant pris des vacances dans le sud de la Corse entre 2011 et 2013 en sont revenues contaminées…
Cette histoire a débuté au Laboratoire de Chimie de Coordination du CNRS, à Toulouse, en 1995, à l’initiative du Dr Bernard Meunier. Elle s’est poursuivie grâce à sa constante inventivité, à la confiance qu’il fait aux chercheurs qui travaillent avec lui, et aussi à la fructueuse collaboration avec des parasitologues, tous co-auteurs des publications ici mentionnées.
Annexe
Références
[1] A. Robert et al. J. A. C. S. 1997, 119, 5968; A. Robert et al. Angew. Chem. Int. Ed. 2001, 1954; A. Robert et al. Acc. Chem. Res. 2002, 35, 167; S. Laurent et al. Angew. Chem. Int. Ed. 2005, 44, 2060.
[2] A. Robert et al. PNAS, 2005, 102, 31676.
[3] C. Loup et al. Antimicrob. Agents Chemother. 2007, 51, 3768; B. Meunier et al. Acc. Chem. Res. 2010, 43, 1444.
[4] B. Meunier, Acc. Chem. Res. 2008, 41, 69.
[5] a) O. Dechy-Cabaret et al. ChemBioChem. 2000, 1, 281 ; b) O. Dechy-Cabaret et al. Chem. Eur. J. 2004, 10, 1625 ; c) O. Dechy-Cabaret et al. brevet CNRS (2000); d) Robert et al. Acc. Chem. Res. 2002, 35,167 ; e) Meunier et al. Acc. Chem. Res. 2010, 43, 1444.
[6] F. Bousejra-El Garah et al. Eur. J. Inorg. Chem. 2008, 2133 ; F. Bousejra-El Garah et al. Antimicrob. Agents Chemother. 2008, 52, 2966.
[7] F. Coslédan et al. PNAS 2008, 105, 17579.
[8] Revue récente sur la chimiothérapie de la schistosomiase : S. Thétiot-Laurent et al., Angew. Chem. Int. Ed. 2013, 52, 7936.
[9] J. Boissier et al., Antimicrob. Agents Chemother. 2009, 53, 4903 ; V. Pradines et al., Antimicrob. Agents Chemother. 2011, 55, 2403 ; J. Portela et al., PLoS Neglected Dis. 2012, 6, e1474.