

Cet article fait partie du dossier présentant le développement chimique, branche de l'industrie pharmaceutique qui participe au développement de la synthèse des principes actifs des médicaments et produit les lots nécessaires aux études cliniques avant commercialisation.
Le premier article rappelle les différentes phases de la « vie » d'un médicament, de la découverte du principe actif à la commercialisation.
Le deuxième article (ci-dessous) décrit les missions des équipes travaillant au développement chimique.
Le troisième article traite du scale-up, autrement dit la transposition à grande échelle d'une synthèse élaborée en laboratoire.
Un glossaire, commun aux trois articles, est accessible à la fin de chaque article.
Les missions du développement chimique
On peut schématiquement découper les tâches incombant aux chimistes de développement selon trois phases, liées aux phases de développement du médicament :
- dans la première, le but est de produire rapidement du principe actif (PA)1 ;
- dans la deuxième, il s’agit de développer des synthèses industrialisables ;
- dans la dernière phase, il s’agit d’optimiser les voies de synthèse retenues à l’issue de la phase précédente.
Être en capacité de produire du principe actif (PA) pour les phases précliniques
L’une des missions premières du développement chimique est de fournir du PA pour les études précliniques2. Les quantités synthétisées sont de l’ordre de la centaine de grammes à quelques kilos : pour cela, un « kilo-lab », constitué par des réacteurs de capacité comprise entre 2 et 30 litres (Figure 1), est utilisé.

Pour des besoins plus importants, un outil de production semi-industrielle, appelé « pilote », dans lequel les réacteurs ont une capacité de 50 à 1 000 litres, est utilisé (Figure 2).

On a vu que les molécules étudiées en phase préclinique ont statistiquement peu de chance d’aboutir au médicament, c’est pourquoi il n’est pas nécessaire d’investir trop d’effort tout de suite. À ce stade, l’objectif principal est d’être capable de produire les premières quantités de produit sans chercher l’optimum et de livrer à temps pour ne pas prendre de retard sur le développement (time to market).
L’aménagement des modes opératoires de la chimie médicinale
Le délai serré implique l’aménagement de la voie de synthèse proposée par la chimie médicinale. Cette voie de synthèse n’est pas toujours industrialisable en l’état, et ce pour plusieurs raisons
- La considération sécurité (HSE) : la sécurité est au cœur de toutes réflexions et de tous les travaux. En tant qu’entreprise responsable, le premier réflexe est de substituer tous les produits Cancérigènes-Mutagènes-Reprotoxiques (CMR) du protocole de synthèse proposé par l’équipe de chimie médicinale au profit de réactifs qui ont une toxicité faible ou nulle. Le but est bien sûr de protéger les personnes et l’écosystème environnant. Manipuler des litres de solvant CMR implique un niveau de sécurité différent de la manipulation de quelques millilitres.
- La productivité : les modes opératoires de chimie médicinale sont très peu productifs et c’est normal, leur but est de synthétiser la molécule, quel qu’en soit le coût. Il est fréquent d’avoir des modes opératoires avec des dilutions importantes pour la réaction et le traitement (entre 60 L/kg et 120 L/kg). Si cela n’est pas problématique pour un essai en laboratoire sur 2 grammes, cela le devient lors des fabrications de 1 à 10 kg où la taille des équipements requis est limitante. Concentrer le milieu réactionnel nécessite forcément une étude de la réaction mise en jeu, car tous les chimistes le savent, la chimie est souvent capricieuse !
- La compatibilité avec les outils industriels : une réaction à l’échelle du laboratoire et une réaction à l’échelle du pilote ne s’appréhendent pas de la même manière. À grande échelle, une mise à sec en fin de traitement ou une chromatographie peuvent être envisagées mais restent à éviter. Sécher une phase organique avec du sulfate de magnésium MgSO4 anhydre est un automatisme de laboratoire qui s’avère rédhibitoire au pilote (du fait d’une quantité de déchet considérable!).
Vous pourrez trouver également d’autres informations dans le 3e article du dossier.
Le lead time (temps de fabrication)
Comme vous l’avez compris, celui-ci doit être le plus court possible. Généralement, pour des synthèses totales de 10 à 15 stades, celui-ci est de quelques mois en incluant :
- la recherche de fournisseurs (sourcing), l’achat et la livraison des matières premières : acheter 10 grammes dans un catalogue de réactifs est commode mais cela est irréalisable pour la commande de dizaines de kilos ;
- l’aménagement de la synthèse ;
- l’analyse et la livraison du lot.
La qualité du PA
Lors des phases précliniques, une pureté supérieure à 99,9% du PA n’est pas nécessaire. Si des impuretés sont présentes, elles sont alors qualifiées d’un point de vue toxicologique : il suffit de s’assurer que la moindre trace d’impureté soit sans danger pour le futur patient.
En revanche, les lots cliniques1 doivent être plus purs et chaque nouvelle impureté doit être caractérisée et sa toxicité étudiée.
Définir les voies de synthèses et développer les modes opératoires lors des phases cliniques
Lorsqu’une molécule passe le cap des tests toxicologiques avec succès, elle continue son chemin et doit prouver son efficacité sur les patients sans montrer d’effets indésirables. Le besoin en PA est alors décuplé et une nouvelle phase du métier se profile. Cette étape se situe en milieu, voire fin de phase II. En moyenne, celle-ci dure plusieurs années et utilise une grande quantité de ressources humaines. Il s’agit du cœur de l’activité d’un projet qui se rapproche de l’industrialisation.
Il devient maintenant impératif de développer une voie de synthèse industrialisable : elle doit être robuste (le rendement et la qualité varient peu lors d’un changement de conditions opératoires), reproductible, compétitive et à faible impact environnemental.
Ceci constitue le cœur du métier, dans lequel la créativité les chimistes de développement est mise à contribution. Le but est désormais d’envisager toutes les rétrosynthèses possibles pour le PA, pour trouver celle qui permettra la production à l’échelle de la tonne. C’est là que les connaissances théoriques et la créativité de chaque chimiste interviennent. Rester à jour sur les avancées de la chimie organique via la littérature scientifique est primordial.
Sur quels critères se base-t-on pour concevoir et choisir une nouvelle voie de synthèse ? Après avoir envisagé toutes les voies de synthèse possibles, il faut les évaluer pour choisir celle qui deviendra l’« élue » : la synthèse qui permettra de fournir du PA à l’échelle de la tonne.
Les grands principes
Le choix de la route définitive se fera selon plusieurs critères, regroupés sous l’acronyme SELECT par leur inventeur à AstraZeneca, GlaxoSmithKline et Pfizer (Chem. Rev. 2006, 106, 3002) :
| Sécurité : manipuler des produits chimiques est dangereux et des accidents peuvent très vite arriver par méconnaissance des risques. On ne peut le répéter assez, la sécurité est au cœur de nos réflexions. Vous trouverez plus d’informations dans le 3e article. |  |
| Environnement : c’est un facteur tout aussi important que la sécurité ! Il est impératif de limiter au maximum notre impact sur l’environnement en substituant les solvants dangereux par des solvants verts (chimie verte). L’augmentation de la productivité volumique permet également de réduire l’impact environnemental et la gestion des déchets ! Des indicateurs comme le facteur-E* (rapport massique déchet/produit désiré) permettent d’évaluer le côté « vert » de nos synthèses. |  |
| Loi : le développement et la commercialisation doit se faire dans le respect des lois relatives à la sécurité et à la propriété intellectuelle en vigueur. Le développement doit également être réalisé en accord avec la réglementation sur certains produits chimiques dangereux (drogues, armes chimiques, explosifs, etc.). |  |
| Economie : mettre sur le marché un médicament présente un coût très élevé, de l’ordre d’un milliard d’euros. Pour contrôler les coûts, il est nécessaire d’avoir le procédé le plus efficient en termes de rendement, de coût des matières premières, de productivité et de robustesse). |  |
| Contrôle : étape importante à l’échelle industrielle, le contrôle de la synthèse et de la qualité du produit obtenu. Il est important de définir des spécifications2 sur les matières premières et intermédiaires pour garantir un produit conforme et éviter des coûts de non-qualité liés à un retraitement du principe actif. |  |
| Throughput (Productivité) : Cet item est lié à l’économie. Il s’agit d’obtenir des synthèses avec le moins d’étapes chimiques possibles, des temps de fabrication courts et des solutions concentrées pour fabriquer le maximum de produit dans un réacteur ! |  |
Dans le détail…
En pratique, pour se conformer à ces cinq grands principes, il faut étudier :
- le prix et disponibilité des matières premières (MP) ;
- le nombre d’étapes de chaque voie ;
- la convergence de la synthèse (voir ci-après) ;
- la faisabilité de la synthèse (analyse de risque) ;
- l’économie d’atome (Trost, B. M. Science 1991, 254, 1471) ;
- le rendement ;
- le facteur-E (environnemental) ;
- l’aspect HSE (réaction exothermique, toxicité etc.).
Il n’est pas toujours possible d’optimiser tous ces critères ; tout est une question de compromis : on pourra par exemple accepter plus facilement une synthèse comprenant une étape de plus mais partant d’une matière première 20 fois moins chère. Par ailleurs, une rétrosynthèse très créative sur le papier mais sans références bibliographiques sera testée uniquement en seconde intention. Si des réactifs, solvants ou matières premières sont chers, il faudra en tenir compte et introduire ces composés le plus tard possibles dans la synthèse.
L’importance de la convergence d’une synthèse !
La synthèse convergente est préférable car elle présente moins de risque et est plus productive qu’une synthèse linéaire (Figure 3).
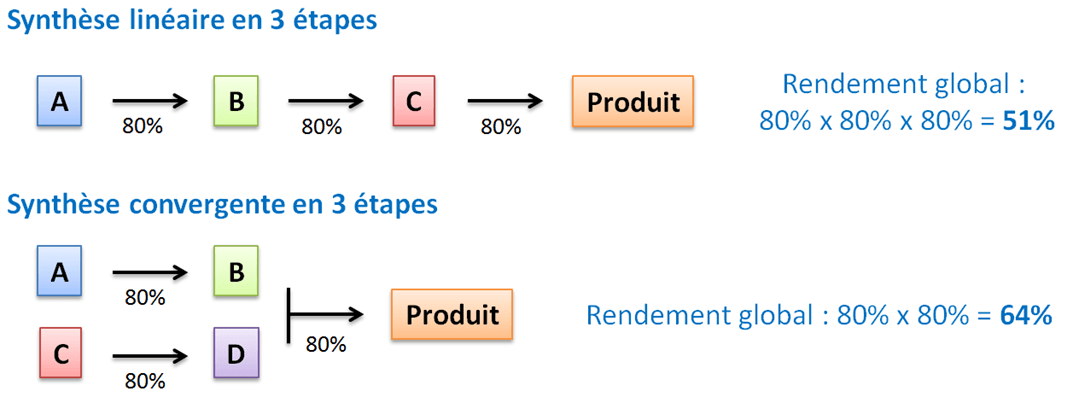
Fabriquer en parallèle des intermédiaires d’une même synthèse convergente fait gagner un temps non négligeable de production. Une synthèse convergente offre également d’autres avantages : d’une part, le rendement global est meilleur ; d’autre part, si l’un des intermédiaires d’une branche est malencontreusement perdu (à cause d’une réaction qui n’a pas marché ou du fait d’erreurs humaines…) il faudra alors moins d’étapes chimiques pour le re-synthétiser.
Optimiser les voies définitives
Ce travail a lieu dans la dernière partie des années de développement, lorsqu’une molécule se trouve en début de phase III. Pour des raisons économiques, gagner un point de rendement supplémentaire est très important et de nombreux efforts de recherche sont mis en œuvre. Pour cela, des outils mathématiques innovants sont utilisés, afin de déterminer les conditions expérimentales permettant de maximiser les rendements.
Pour optimiser une réaction, une première approche consiste à faire varier un paramètre à la fois. Cette méthode est appelée OVAT (One Variable at A Time) : on change un paramètre jusqu’à trouver sa valeur optimale, puis on fait varier un autre paramètre en gardant le précédent à la valeur optimale déterminée… et ainsi de suite. Pourtant, cette approche ne permet pas toujours de déterminer les conditions opératoires optimales. Le diagramme (Figure 4) ci-dessous parle de lui-même :
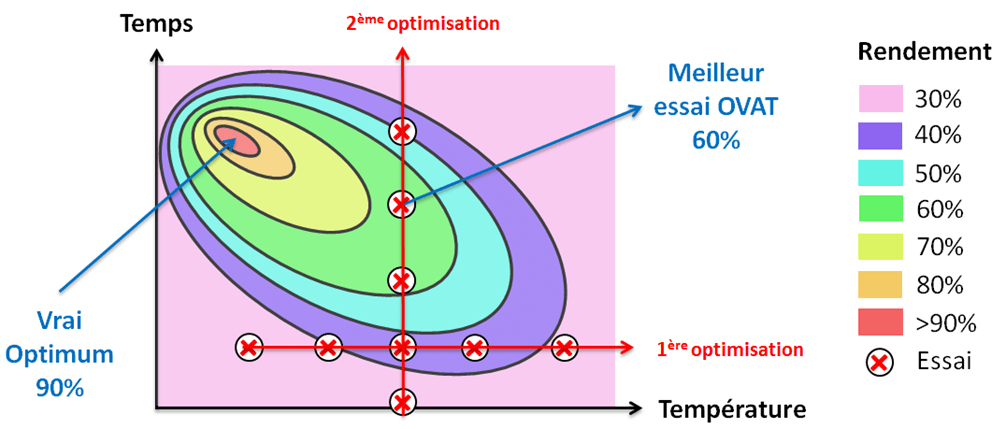
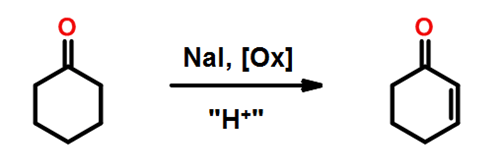
On voit clairement que la méthode OVAT ne permet pas toujours de trouver le point qui donnera le meilleur rendement… à moins que la chance s’en mêle. L’erreur que l’on fait généralement est de penser que chaque paramètre est indépendant des autres dans une réaction. C’est intuitif mais incorrect, il peut exister des interactions importantes entre deux ou plusieurs paramètres. C’est pourquoi nous mettons en œuvre des plans d’expériences, séries d’essais visant à obtenir un maximum d’informations en un minimum d’expériences. Bien réalisés, ces plans nous permettent de trouver le point optimum dans un espace expérimental défini par l’expérimentateur grâce à une modélisation mathématique des paramètres étudiés.
Prenons un exemple : lors de l’optimisation d’une réaction d’oxydation (Figure 5) dans laquelle l’iodure de sodium NaI jouait le rôle de catalyseur, le logiciel prédit, après plusieurs essais (dans lesquels plusieurs paramètres ont été modifiés en même temps), que la combinaison des paramètres vitesse d’agitation (puissance fournie, qualité de mélange) et la quantité de NaI influencent fortement le rendement.
Pour étudier deux paramètres lors d’un plan d’optimisation, une dizaine d’essais sont nécessaires. A chaque essai, la masse de produit formé est relevée, l’objectif étant qu’elle soit la plus élevée possible. Voici le graphique (Figure 6) obtenu grâce à ce logiciel appuyé par des essais au laboratoire :
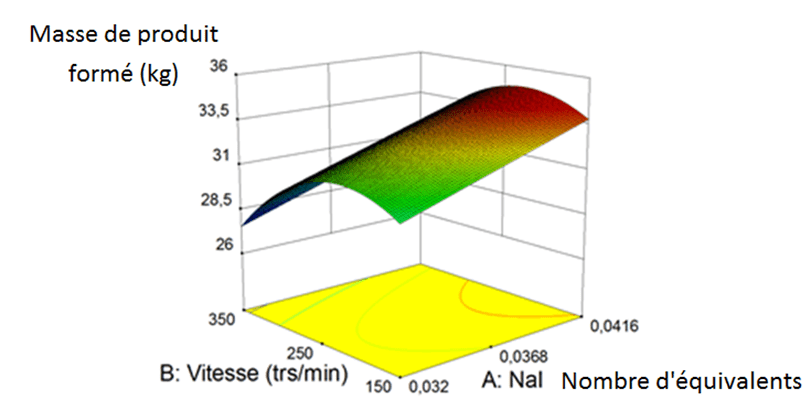
Nous avons pu ainsi déterminer les valeurs des paramètres agitation et quantité de catalyseur nécessaires pour obtenir le rendement maximum. On fait cela sur tous les paramètres influents ou sur la combinaison de ces paramètres, et cela pour chaque étape de la synthèse.
Etape suivante : établir des champs opératoires. Les champs opératoires permettent de définir une plage pour tous les paramètres réactionnels (température, quantités des réactifs, etc.) dans laquelle un écart n’aura pas d’impact critique sur la qualité du produit.
Côté théorique, il est nécessaire de bien comprendre les réactions mises en jeu car toutes les impuretés dont la teneur est supérieure à 0,1 % doivent être caractérisées. Ensuite, elles sont synthétisées à l’échelle de la dizaine de gramme pour évaluer leur toxicité et ajuster les spécifications de celles-ci dans le PA.
Appuyer les affaires réglementaires
L’aspect réglementaire de notre travail est très important, il est pris en compte dès la production des lots pour les études cliniques. Toutes les productions se font selon les Bonnes Pratiques de Fabrication (BPF, ou GMP pour Good Manufacturing Practice en Anglais). C’est une démarche de qualité nécessaire qui est inspectable par les autorités compétentes. Ces autorités sont des organismes de droit public, la FDA (Food and Drug Administration) pour les Etats-Unis, l’EMEA (Agence Européenne du Médicament) pour l’Europe et l’ANSM (Agence Nationale de Sécurité du Médicament) pour la France. Ils analyseront minutieusement la production des lots pour vérifier leur traçabilité et la conformité des spécifications définies dans l’AMM1 ou dans l’IMPD (Investigational Medicinal Product Dossier), le but étant d’écarter tout risque pour le patient qui remettrait en cause la qualité, l’efficacité et la sécurité du produit.
L’IMPD est un dossier réglementaire à fournir pour pouvoir réaliser une étude clinique et l’AMM est le dossier réglementaire qui permet d’obtenir l’accord de mise sur le marché d’un médicament. Une partie du travail des ingénieurs de recherche en développement chimique consiste à participer à l’écriture de ces différents dossiers.




