Le collage est l'une des premières techniques d'assemblage d'une structure que l'Homme ait utilisée en complément éventuel de celle du coincement pour l'emmanchement. Une autre technique, toute aussi ancienne, consistait à réaliser une ligature sur un manche en bois, en os ou encore en corne, les liens étant généralement des tendons de cervidés.
Les différents types d'assemblages au cours des âges
L'assemblage, entrepris dès la Préhistoire, consiste en l'opération élémentaire de faire en sorte que deux objets soient rendus solidaires. Cela implique la réalisation de formes stables obtenues par la réunion appropriée de plusieurs autres formes réalisées indépendamment et par des procédés qui peuvent être différents. On peut dénombrer six principaux types d'assemblages :
L'assemblage statique par juxtaposition
Il concerne principalement la construction. Mis à part le cas banal de murs de pierres de taille assemblées à sec, il n'en est plus de même pour la voûte, d'origine grecque, considérablement développée par les Romains, ni celui de la coupole dont l'invention est romaine. Entrent également dans cette catégorie, les arcs d'ogives et les contreforts de la construction médiévale.
L'assemblage par blocage géométrique
Assemblage par emboîtements exacts de pièces de matériaux divers (pierres de taille, bois, fer, etc.) par l'association d'une excroissance et d'une cavité utilisés dès l'Antiquité classique (assemblages de colonnes, de balustrades, de conduits en poterie ou en fonte) avec éventuellement l'aide d'une pièce auxiliaire, "double queue d'aronde" pour les pierres de taille, "cheville" pour la technique tenon et mortaise essentiellement dans le bois ou encore "clavette" pour le métal, ce qui suppose la maîtrise du perçage et du forage, lesquelles remontent au néolithique.
L'assemblage à force
Type d'assemblage dont relève le "clou", utilisé dès l'âge du fer puis très développé par les Romains pour l'assemblage de pièces de bois. L'apparition de la "vis à bois" est beaucoup tardive (Moyen-Âge), quant à la "vis à fer", elle n'apparaît qu'au XVIIIe siècle, l'assemblage nécessitant en effet la réalisation délicate de deux filetages de pas et de diamètre constants s'ajustant exactement. Il en est de même du système "vis-écrou", dont l'ébauche remonte à l'Antiquité romaine. Dans le même genre d'assemblage à force entre le "rivetage" qui a été réalisé dès l'âge des métaux, mais de façon grossière. Il s'est essentiellement développé et n'est devenu systématique, étant alors mécanisé, qu'au XVIIIe siècle pour les assemblages de plaques de fer.
L'assemblage de fils
Assemblage qui consiste soit à "l'entrecroisement" de plusieurs fils, pour réaliser un "tissage" par exemple, dont la technique remonte au néolithique, soit au "maillage" d'un seul fil, comme dans le "tricot", lequel n'apparaît qu'à partir du Moyen Âge. Ces deux types d'assemblages ont connu une mécanisation progressive qui a atteint un très haut degré de technicité à la fin du XVIIIe siècle en Grande-Bretagne et près d'un demi-siècle plus tard en France.
L'assemblage de surfaces souples
Assemblage qui est connu dès la Préhistoire pour réunir des pièces de tissus et également de cuir. On différencie traditionnellement d'une part, les assemblages amovibles à l'aide d'une "épingle", d'abord réalisée en corne, en bois, en os puis en métal, d'une "fibule" (actuelle "épingle à nourrice", dite de sûreté), d'une "agrafe", d'un "bouton" (technique qui ne s'est généralisée qu'au Moyen Âge), d'un "bouton-pression" qui ne date que du XVIIIe siècle, et, d'autre part, les assemblages fixes par le "bâti" et la "couture" des tissus. Cette technique suppose l'existence de "l'aiguille à chas" qui est inventée à la fin de la Préhistoire. La mécanisation de la couture est assez tardive puisque la machine à coudre n'apparaît qu'à la fin du XIXe siècle.
L'assemblage par adhésion
Les "colles", apparaissent excessivement tôt dans l'histoire des techniques pour l'assemblage de matériaux très divers. Aujourd'hui, on les utilise pour réunir deux surfaces, pour assurer un joint étanche ou encore pour relier des particules ou des fibres dans les matériaux composites. Il faut également citer la "soudure", que l'on confond souvent avec le collage, laquelle ne porte au départ que sur les métaux, elle s'est peu à peu substituée à l'assemblage par rivets et par boulons.
Historique du collage
Généralités
Le collage est l'une des premières techniques d'assemblage d'une structure que l'Homme ait utilisée en complément éventuel de celle du coincement pour l'emmanchement. Une autre technique, toute aussi ancienne, consistait à réaliser une ligature sur un manche en bois, en os ou encore en corne, les liens étant généralement des tendons de cervidés.
L'attrait pour le collage n'est pas fortuit, il faut signaler que la nature a toujours été particulièrement généreuse, puisque de tout temps et en tout lieu, elle a mis à la disposition de l'Homme un grand nombre de produits adhésifs naturels d'origines végétale, animale et minérale tels que la glu extraite de l'écorce tendre du houx, la chair visqueuse du fruit du gui (utilisée par les Romains pour piéger les petits oiseaux), le jus de gousse d'ail pressée (employé au début du XXe siècle par les lunetiers pour parfaire le maintien des verres dans leur monture), la gomme arabique extraite de plusieurs variétés d'acacias, comme l'Acacia sénégal, appelé aussi gommier, (la gomme arabique est le principal composant de la pastille Valda®, qui, comme on le sait, colle aux dents et est tout à fait capable de provoquer le descellement non souhaité d'une prothèse dentaire), le latex tiré de certaines espèces d'hévéas dont on tire le caoutchouc, comme base de nombreuse colles, la sève résineuse des conifères, les farines de céréales, le goudron de charbon de bois, la bétuline (brai de bouleau obtenu par calcination à l'étouffée de l'écorce de l'arbre, qui servit pour réparer des céramiques brisées et coller les lames en silex des premiers outils en Occident), les blanc et jaune d'oeuf ainsi que le miel (utilisés au Moyen Âge et au début de la Renaissance pour lier les pigments colorés et coller les feuilles d'or sur les manuscrits enluminés), la gomme laque sécrétée par certaines espèces de cochenilles, la cire d'abeille (employée pour imperméabiliser les premières céramiques et pour les sceaux), la caséine du lait (utilisée au début XXe siècle dans l'aéronautique naissante), le bitume (substance naturelle minérale fossile que l'on trouve généralement dans les bassins sédimentaires) employé très tôt en parallèle avec la bétuline pour coller les premiers outils), etc., les exemples ne manquent pas.
Les collages chez nos amis les bêtes
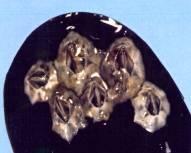
Avant l'Homme, certaines espèces animales ont utilisé et emploient encore des techniques de collage pour assurer leur survie. À titre d'exemples, citons les araignées qui enduisent certains fils de leur toile afin de piéger les insectes dont elles se nourrissent. Signalons également le cas des moules qui ont résolu leur problème de fixation, sur les rochers et sur les bouchots, dans des conditions que nous qualifions aujourd'hui de technologiquement difficiles, à savoir en milieu aqueux et salin : le byssus est composé d'une protéine sécrétée par l'animal, laquelle s'organise en un faisceau de filaments soyeux qui adhèrent aux supports par des interactions électrostatiques. À l'heure actuelle, on sait préparer par génie génétique la protéine et les milieux médicaux et dentaires sont très attentifs aux solutions qu'est susceptible d'apporter cette protéine aux problèmes d'adhésion et de suture en chirurgie. Parmi les nombreux invertébrés marins "collés", citons les balanes (figure 1) qui sont des petits crustacés pyramidaux de 2 à 3 mm de haut, que l'on trouve sur les coquilles de moules et d'huîtres, sur les rochers et malheureusement aussi sur les coques des bateaux au grand dam des plaisanciers, dont la fixation est assurée à l'aide d'un ciment résultant de la polymérisation d'éléments moléculaires (heptapeptides) par l'intervention d'une enzyme appelé phénol-oxydase, sécrétés par des glandes spécialisées de l'animal.
Une étude récente entreprise aux USA, entreprise par Autumn et ses collègues de l'Université de Berkeley [1], fait apparaître que le gecko (figure 2), un lézard qui vit dans les régions tropicales, est capable de se mouvoir sur n'importe quelle surface lisse verticale ou sous un plan horizontal tout aussi lisse, par la seule action des forces de Van der Waals. Baptisées du nom d'un physicien hollandais de la fin du XIXe siècle, ces forces sont des interactions de très courte portée qui naissent des phénomènes d'attraction électromagnétique, liés aux fluctuations extrêmement rapides des distributions électroniques autour des noyaux atomiques. Les pattes du gecko (qui ne porte pas de griffes comme ses cousins de nos pays tempérés) sont terminées par cinq doigts dont l'observation au microscope électronique à balayage fait apparaître qu'ils présentent chacun environ 5000 poils de kératine par millimètre carré, qui se divisent à leur terminaison en plusieurs centaines de soies (figure 2). Au total, ce lézard possède environ deux milliards de soies qui lui assurent à la fois suspension et progression. Si l'on suppose que chaque liaison crée une force d'attraction de l'ordre de 20 nanonewtons conformément aux estimations récentes, le lézard peut ainsi supporter une masse de 4 kilogrammes, alors qu'il ne pèse lui-même pas plus de 200 à 300 grammes. Le problème est de trouver le moyen (harnais, ceinture) d'accrocher le poids afin de vérifier l'estimation !
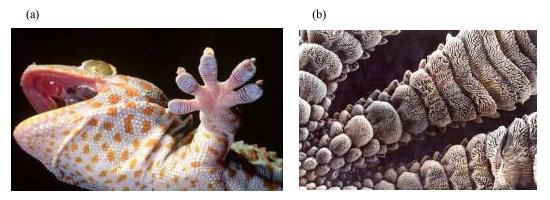
Il ne s'agit pas ici, à proprement parler, de collage puisqu'il n'y a pas intervention d'un troisième corps. On cite souvent cet exemple car les forces de Van der Waals interviennent de manière prépondérante dans les phénomènes d'adhésion. On pense que la technologie "naturelle" utilisée par le gecko permettrait de réaliser des nanosutures chirurgicales. D'ailleurs, une étude récente menée par Geim [2], a permis de réaliser à l'aide de polyimide, un polymère synthétisé dès la fin des années 1960, un ruban adhésif comportant des poils de 2 micromètres de long environ qui jouent le même rôle que les soies de kératine du gecko (figure 3). Ce ruban pourrait être utilisé également pour la préhension de nano-structures.

Les forces de surface
Pour expliquer l'existence des forces de surface, rappelons que la cohésion d'un corps est assurée par des forces qui agissent en son sein et chaque atome est soumis à des interactions avec ses plus proches voisins de sorte que la résultante des interactions est nulle. Par contre, à la surface, les atomes se trouvent soumis à une force résultante, non nulle, dirigée vers l'intérieur et qui tend à réduire la surface. C'est cette tension, dite superficielle et qui s'exprime en newton par mètre, qui fait que les bulles de savon sont parfaitement sphériques et qu'il en est de même pour les gouttes qui sont en apesanteur. Du fait de l'existence de la résultante des forces, les molécules de l'atmosphère environnante sont attirées par la surface du solide et viennent la polluer. En fait, il existe un équilibre statistique entre la condensation des molécules qui se déposent et l'évaporation des molécules qui quittent la surface.
Les forces d'adhésion sont de même nature que celles qui assurent la cohésion des solides : les matériaux métalliques et covalents ont des énergies superficielles (énergies libres de surface) très élevées, de l'ordre de 1 à 3 J.m-2, pour les cristaux ioniques la gamme d'énergies s'étend de 100 à 500 mJ.m-2, quant aux matériaux moléculaires, leurs énergies superficielles sont inférieures à 100 mJ.m-2, ordre de grandeur de la liaison hydrogène et des forces de Van der Waals. Ces dernières ont été étudiées par les compatriotes de Johannes Diderik Van der Waals, Wilhelm Keesom et Petrus Debye. Le premier, en 1912, postulait que les molécules s'attiraient à l'image de petits aimants (figure 4) sous l'effet de forces d'attraction. Effectivement, une molécule dont les centres de gravité des charges positives et des charges négatives ne sont pas confondus est assimilable à un dipôle.
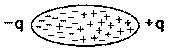
Ce modèle, qui ne convenait pas aux molécules non polaires, a été amélioré par le second (Debye) en 1920 en expliquant qu'il suffisait qu'une molécule soit polaire et attirait les autres par influence, en créant un dipôle induit, sous l'effet précisément de forces d'induction. L'américain Fritz London, en 1930, a montré que l'attraction des molécules non polaires résulte des interactions électromagnétiques produites par les fluctuations rapides continuelles dans la distribution des électrons à l'intérieur des molécules, qui font qu'à chaque instant la molécule se comporte comme un dipôle de très courte durée de vie, et les molécules s'attirent sous l'action de forces dites de dispersion. La fréquence de la modification de la configuration électronique est de l'ordre de 1015 hertz. Enfin, la théorie généralisée a été proposée par Lifshitz en 1955 en faisant intervenir les constantes diélectriques des milieux considérés.
Pourquoi donne-t-on tant d'importance aux forces de van der Waals ? Tout simplement parce que, dans une atmosphère ordinaire, l'air ambiant par exemple, les forces les plus intenses, qui sont à très court rayon d'action, sont écrantées par les films d'oxydes, les couches de molécules adsorbées et toute contamination extérieure présents sur la surface exposée de sorte que la contribution des forces de Van der Waals, dans cette situation, devient prépondérante. Bien évidemment, si l'on élimine la contamination atmosphérique, les couches d'oxydes peuvent alors réagir par des liaisons polaires coordinantes, appelées aussi liaisons accepteur-donneur (sous-entendu d'électrons) ou liaisons acide-base ou encore forces de Lewis, ou par des liaisons ioniques plus intenses.
Chronologie des colles et des techniques utilisées par l'Homme avant l'ère chrétienne
Les premières manifestations de l'utilisation par l'Homme de la viscosité et de la plasticité conférant aux matières naturelles leur pouvoir adhésif remontent à environ 3500 ans avant le début de l'ère chrétienne, par la découverte dans le désert de Judée, dans l'actuelle Cisjordanie, ainsi qu'en Basse Égypte, d'instruments ressemblant à des faucilles réalisées en bois armées d'éclats de silex maintenus par du bitume. Dans le désert, le sel ronge les manches en bois et il ne subsiste le plus souvent que le silex collé au bitume (figure 5). Heureusement parfois, le sol sablonneux a conservé l'empreinte du manche, ce qui permet d'affirmer qu'il s'agit bien d'éléments de faucilles.

Au Proche-Orient, la première colle utilisée par l'homme fut sans conteste le bitume, et cela depuis les temps immémoriaux, comme cela sera vu ultérieurement. D'origine minérale et fossile, comme le pétrole, le bitume affleure dans les bassins sédimentaires. En Occident, la première colle utilisée est semble-t-il la bétuline, c'est-à-dire le brai de bouleau, que l'on obtient par calcination à l'étouffée de l'écorce de l'arbre, comme cela se pratique encore de nos jours en Finlande. Des études récentes [3], par chromatographie en phase gazeuse et par spectrométrie de masse sur des agrégats noirâtres et informes trouvés sur le site paléolithique de Königsaue en Allemagne, ont permis de mettre en évidence la présence de brai de bouleau, qui fut probablement utilisée pour emmancher des pointes de flèches et réparer des poteries endommagées et également les rendre imperméable aux liquides.
Sur le site sous-lacustre d'Auvernier, près du lac de Neuchâtel en Suisse, appartenant à la culture de Cortaillod, on a découvert une espèce de couteau à moissonner (figure 6), avec une lame de silex isolée d'une longueur d'une dizaine de centimètres, coincée dans une rainure ménagée dans le manche en bois d'if et maintenue en place à l'aide de bétuline, datant d'environ 3700 ans avant notre ère [4].

En France, des fouilles entreprises dans les stations lacustres des lacs de Clairvaux et de Chalain, dans le Jura, ont permis de mettre à jour un certain nombre d'armes et d'outils utilisés par les palaffiteurs, tels que des haches de pierre avec leurs manches auxquels elles étaient collées à l'aide d'un mastic résineux.
En Suisse, près de Fribourg, dans le gisement sous-lacustre de Montilier, on a découvert divers objets dont des couteaux à moissonner type "Horgen" (figure 7), pour lesquels le collage sur le manche en bois est assuré par de la bétuline. La forme arrondie des lames fait que les archéologues pensent que ces ancêtres du "couteau suisse" pouvaient également servir de hachoir. Ces objets sont datés 3150 ans avant notre ère. L'excellent état de conservation des manches en bois résulte d'un effet d'embaumement par les plantes des cités lacustres dû à la combinaison chimique des constituants des végétaux avec l'eau.

Une faucille néolithique découverte en Turquie et conservée à Ankara, montre très clairement comment le bitume permettait de maintenir des lames de silex dentelées sur un manche en bois (figure 8). On a également retrouvé des faucilles égyptiennes quasi identiques, plus récentes, datant environ de 1400 ans avant l'ère chrétienne, actuellement conservées au British Museum à Londres.

La fabrication du premier outil est manifestement plus ancienne. En effet, la découverte récente en Syrie de silex recouverts partiellement de bitume (figure 9a) et concomitamment le déchiffrage de textes cunéiformes sur des plaquettes d'argile, laissent à penser à la fabrication du tribulum à la fin du Néolithique au Proche-Orient, soit juste après la domestication animale, c'est-à-dire il y a 8000 ans [5]. Le tribulum est une espèce de traîneau lesté, tel que la représentation d'artiste le montre dans la figure 9b, constitué de rondins de bois assemblés par des ligatures et entre lesquels sont insérés et collés des silex, dont le rôle est de séparer le grain de la paille après fauchage, permettant ainsi de nourrir à la fois l'homme et l'animal.

Le tribulum est toujours utilisé actuellement en Syrie, mais dans une forme moderne : le traîneau est constitué de la tôle aplanie d'un gros bidon dans laquelle on a ménagé des accrocs acérés jouant le rôle des silex d'antan. Voilà un bel exemple de la récupération moderne des techniques ancestrales (figure 10).

En fait, la découverte en 1996, également en Syrie, sur le site de Umm el Tlel, de silex grossièrement taillés sur lesquels on observe des traces de bitume (figure 11), datant d'il y a au moins 50000 ans (l'époque moustérienne, c'est-à-dire le paléolithique moyen, autrement dit l'ère néandertalienne), est susceptible de faire reculer très loin dans le temps les premières fabrication et utilisation d'un outil collé [6].

Aux époques les plus reculées, le collage n'était pas uniquement réservé à la réalisation d'assemblages devant supporter des efforts mécaniques. Cette technique fut très tôt utilisée pour maintenir en place sur un support non noble, comme le bois, un revêtement décoratif. Ainsi, au cours de la période Chalcolithique ou l'Uruk, au quatrième millénaire avant Jésus-Christ, les artistes mésopotamiens créaient de remarquables compositions, réunissant des pierres de couleur bleue (lapis-lazuli) et rose (tourmaline de type rubellite), de la nacre, des coquilles et des fragments d'os sculptés et gravés, harmonieusement maintenues en place sur une âme, en matériau banal tel que le bois, recouverte de bitume. A Suse, dans l'actuel Iran, un sondage pratiqué dans les années 1930 sous l'acropole a permis de découvrir un magnifique lion en bois, dont l'âge est estimé à 3300 ans avant notre ère, recouvert d'une plaque en or maintenue sur la surface du bois à l'aide de bitume utilisé comme liant intermédiaire.
Des documents égyptiens remontant à quelque 10 siècles avant l'ère chrétienne portent témoignage de la pratique du premier collage par fusion ("Hot Melt" actuel, dans le langage anglo-saxon), lequel était utilisé pour assembler des lames et des pointes de flèches dans leurs manches et leurs tiges respectifs à l'aide de soufre. Enduits de soufre, les objets coupants et pointus étaient mis en place par emmanchement puis chauffés à une température, que l'on sait aujourd'hui être égale à 115 °C, afin de provoquer la fusion du soufre, liant qui, après refroidissement, assurait la cohésion de l'assemblage souhaité. Plus tard, selon le même principe, les Romains utiliseront le plomb. Ces mêmes Romains mirent au point, sans doute par jeu et aussi très certainement pour agrémenter leurs repas, le collage par contact dont firent les frais les petits oiseaux qui venaient se poser sur des planchettes enduites de la chair visqueuse du fruit du gui et y restaient piégés.
Phéniciens et Romains, pour coller le bois, utilisaient fréquemment des os écrasés et des déchets de poissons pour fabriquer des colles d'origines animales. Ce type de colles ainsi que des résines de conifères, mélangé d'ocre, de calcaire et de différentes terres broyées était couramment utilisé dans l'antique Égypte pour décorer les cuves et couvercles des sarcophages. Il s'agit bien là d'une réelle opération de collage, puisque l'adhésif a pour effet de fixer les pigments de diverses couleurs sur un substrat.
Tout comme le pratiquent encore les pêcheurs européens qui rendent étanches les assemblages de planches en bois constitutives des coques de leurs bateaux à l'aide d'un mélange de chanvre, de sisal et de poix, les Assyriens et les Phéniciens, calfataient les bateaux à l'aide de compositions, à base de térébenthine (extraite du pistachier résineux appelé térébinthe), de cire d'abeille, de goudron de charbon de bois et de bitume, lesquelles présentaient un excellent comportement à l'humidité (figure 12) [7].

La cire d'abeille, de couleur naturelle ou diversement pigmentée à l'aide de terres finement broyées, fut depuis la plus haute Antiquité utilisée pour les sceaux, technique qui permettait à la fois de fermer très symboliquement un pli et de graver par moulage une signature identifiant sans conteste l'expéditeur, à l'aide d'un corps dur gravé (pierre ou métal) formant matrice plane ou cylindrique. Cette technique est toujours utilisée pour certains documents officiels et pour les scellés divers. Mais dans le quotidien d'aujourd'hui, le ruban adhésif vient à notre secours pour parfaire la fermeture d'un pli. Des études chromatographiques récentes [3] ont montré qu'il existe une analogie frappante (figure 13) entre la cire d'abeilles actuelle et celle retrouvée sur les parois internes de tessons de céramiques de l'époque néolithique, cire vraisemblablement utilisée pour imperméabiliser ces récipients devant contenir des liquides.
![Chromatogrammes partiels d'une cire d'abeilles actuelle [en haut] et d'un échantillon archéologique du site de Bercy [en bas]. (Document : Martine Regert).](/sites/default/files/nodeimages/images/dossiers-structure-macroscopique-article-Collage_Barquins-12.jpg)
Quant à l'utilisation de cire d'abeilles pour les sceaux, la figure 14 montre le sceau-cylindre (a), ainsi que le moulage de son empreinte sur terre glaise cuite (b), du scribe Kalki. Il est réalisé en diorite tachetée, qui est une roche éruptive granitoïde formée de cristaux de feldspath (blanc) et d'amphibole (verte). Haut de 3,3 cm, ce sceau remonte à la Dynastie d'Akkad (Mésopotamie), vieille de 4000 ans [8].

Les anciens Égyptiens pouvaient revendiquer d'avoir trouvé les premiers deux industries majeures, celle de la préservation des aliments et celle des matières plastiques. En effet, on a pu mettre en évidence que les jarres remplies d'épices, découvertes dans la tombe non pillée de Toutankhamon à Thèbes le 4 novembre 1922 par Howard Carter et son mécène Lord George Herbert Carnarvon, étaient hermétiquement scellées avec de l'asphalte fondue, qui est un mélange de calcaire, de silice et de bitume. Ces jarres font partie des 2099 objets répertoriés dans la tombe du célèbre pharaon, mort 1343 ans avant le début de l'ère chrétienne, et remplissent douze salles du musée égyptien du Caire. Rappelons que le masque en or du célèbre pharaon, qui pèse 11 kilogrammes, est recouvert de barrettes de lapis-lazuli et autres pierres précieuses collées à l'aide de bitume.
L'ère chrétienne : début du développement des papyrus et papiers
En Égypte, à partir de la conquête d'Alexandre III, dit le Grand, en 331 avant Jésus-Christ et jusqu'à l'invasion arabe conduite en l'an 641 de notre ère par le général 'Amr Ibn Al-As, une industrie prend toute son ampleur, celle de la cellulose, sous la forme de la préparation des papyrus dont les premiers remontent au XVIIIe siècle avant notre ère. Le papyrus, cet ancêtre du papier, que l'on fabriquait à partir de la moelle d'un roseau, le "roseau d'Egypte" (Cyperus papyrus) qui est un jonc à section triangulaire, ou du cœur; de l'herbe d'une variété du souchet, coupé en bandes, lesquelles étaient placées en croix en deux couches superposées, fibres longues dans un sens et plus courtes dans la direction perpendiculaire pour pouvoir ensuite plus aisément rouler l'ensemble. Mouillées à l'eau du Nil puis séchées au soleil, les bandes étaient ensuite pressées et martelées, le jus exsudé servant de colle, la phase terminale consistant en un polissage fin. Cette industrie très prospère distribuait aux commerces locaux et à l'exportation plusieurs qualités de produit allant du papyrus royal, très fin, réservé à l'écriture, au dessin et à la peinture (figure 15), à une forme beaucoup plus emporétique et grossière destinée à l'emballage. Papyrus détériorés et archives périmées étaient vendus à bas prix aux fabricants de cercueils qui les malaxaient et les agglutinaient pour former une espèce de cartonnage en "papier mâché" dont on enveloppait les momies avant de les recouvrir des traditionnelles inscriptions funéraires. Ainsi, dès cette époque lointaine on pratiquait déjà le recyclage !
Pour ce qui concerne le papier, bien que l'on ait découvert en Chine en 1957, dans une tombe à Baqiao, des boulettes de papier de chanvre ne dépassant pas 10 cm2 (figure 16), dont l'une a pu être datée à un moment compris entre 140 et 87 ans avant le début de l'ère chrétienne (dynastie des Han), l'inventeur officiel est Caï Lun, factotum de l'impératrice Dengsui. En l'an 102 de notre ère, elle lui réclame de quoi écrire, à savoir du papier et de l'encre. Caï Lun, eunuque de surcroît, se met en devoir de répondre à l'injonction de sa souveraine et suggère de fabriquer du papier à partir de l'écorce des arbres, de vieux cordages et chiffons et des filets de pêche détériorés. Il y parvient trois années plus tard, soit en l'an 105. Ce papier était fabriqué feuille à feuille. Comme pour le papyrus, la matière première est végétale, il s'agit de cellulose. Après son premier succès, des matériaux de choix furent utilisés comme des chiffons de lin, de chanvre et de coton, puis ce fut au tour d'autres végétaux, comme les pousses de bambou, la paille de riz et le liber du mûrier blanc et du mûrier à soie, les fibres étant encollées avec un mucilage issu de l'igname. On utilisa également la substance gommeuse localisée sous l'écorce de l'actinidia, l'arbre qui produit le kiwi.

Le papier parvint en Europe, plus précisément en Espagne, en 1154, par le truchement des Arabes et des Turcs. À cette même époque, on colle le bois avec du lait caillé mélangé à de la chaux. Les premières plaques à imprimer, furent bien évidemment chinoises, elles furent réalisées par un certain Bi Sheng en l'an 1040, soit très exactement 400 ans avant Gutenberg ! Elles étaient obtenues par collage sur plaque de fer, à l'aide de résine de conifères, de caractères réalisés en terre glaise cuite.
Au XVe siècle, avec l'essor de la typographie et de l'imprimerie, l'utilisation des colles d'origine végétale et animale s'est accrue. Il faut signaler que la fabrication du papier consomme une énorme quantité de colles : colle de fécule tout d'abord, puis colle de gélatine, beaucoup plus résistante et enfin colle de résine, toute aussi résistante mais moins coûteuse. Le choix de la colle était aussi guidé par les qualités particulières du papier que l'on souhaitait. Par exemple, on constate rapidement que l'ajout de charges minérales à la colle, telles que talc et kaolin, a pour effet d'augmenter l'opacité du papier et d'améliorer son imperméabilité. Sans colle, le papier à lettre serait un véritable buvard !
Quant aux papiers peints d'ameublement, qui sont issus de l'art des imagiers médiévaux, ils étaient ornés, jusqu'au début du XXe siècle, de motifs peints à la colle, d'où leur nom. Après avoir été collés sur le plâtre au moyen d'une colle de pâte obtenue en mélangeant et en chauffant de la farine et de l'eau, on améliora le comportement à l'humidité et aux moisissures concomitantes en dispersant un peu de plâtre au sein de la colle. Dès le XIXe siècle, des collages plus performants encore furent réalisés avec les amidons et les fécules. Plus résistants, ils devenaient également moins sensibles aux bactéries.
S'il est vrai que pendant des siècles la technologie du collage n'a évolué que très lentement, il convient de signaler que malgré une technique parfois très rudimentaire, cette dernière s'est révélée tout à fait remarquable, en menuiserie d'art, en ébénisterie et plus particulièrement en marqueterie et en tabletterie, dans le plaquage de bois à la colle dite abusivement "forte" (colle de peaux, colle de nerfs ou encore colle d'os), permettant ainsi la réalisation au XVIIe siècle d'un très grand nombre d'œuvres d'art que l'on peut encore admirer de nos jours (figure 17). On s'abstient aujourd'hui de parler de colle "forte" pour la raison simple suivante : ou bien le produit est adapté au problème posé et la structure collée va donner entière satisfaction, ou bien, il n'est pas prévu pour l'emploi envisagé et dans ces conditions, on l'abandonne pour un autre.

Les sceaux à la cire d'abeille, pour cacheter les enveloppes, étaient encore couramment utilisés au XVIIIe siècle alors que le collage de simples papiers se faisait à l'aide de bâtonnets à base de gomme arabique, de sucre et d'ingrédients parfumés divers. Le cachetage par sceaux disparut au profit du gommage des enveloppes à l'aide d'un mélange de gomme arabique et de sucre puis à celui, plus économique, de colle d'os, de dextrine et de glucose. Au début du XXe siècle, se développent l'empaquetage ainsi que l'étiquetage, d'abord manuels puis automatiques, à l'aide de papier kraft gommé, le gommage étant à base de sucre. Il n'y a pas si longtemps encore, on collait les étiquettes sur les bouteilles de vin à l'aide de lait, mais la fraude et les contrefaçons font qu'elles sont maintenues aujourd'hui avec un adhésif qui les rendent indécollables sans risque de déchirure.
Le développement des adhésifs synthétiques ne va pas tarder à prendre toute son ampleur, dès le milieu du XIXe siècle, il va nous faire passer du progrès de la technique au progrès technique proprement dit. À titre d'exemple, pour les étiquettes, les timbres et autres objets du même type (comme les chatoyantes "gommettes", à la maternelle), on est passé dans un premier temps du papier à coller (papier peint, affiche), puis au papier préencollé qu'il suffit d'humecter (timbres, feuilles de papier gommé à cigarettes et également papier peint), pour aboutir actuellement au papier auto-adhésif, autrement dit l'autocollant immédiatement prêt à l'emploi après décollement du support à basse énergie de surface qui permet le décollement sans déchirure (enveloppes, macarons publicitaires, coins photo, œillets de renfort, papiers d'ameublement, etc).
De nombreuses colles étant à base de caoutchouc, souvenons-nous que le caoutchouc naturel (le cis-polyisoprène,), issu du latex circulant dans les canaux laticifères de l'écorce interne tendre d'un certain nombre de variétés d'hévéas, était connu des Olmèques qui vivaient voici quelque trois milliers d'années dans la région chaude de l'actuel golfe du Mexique [9]. Le monomère isoprène a d'abord eu comme formule C10H16 proposée dès 1826 par l'anglais Michael Faraday. Il n'était pas loin de compte puisque la formule exacte du monomère isoprène est C5H8, suite à la synthèse entreprise par le chimiste français Gustave Bouchardat en 1879. Le monomère isoprène mesure environ 0.5 nm tandis que la molécule de cis-polyisoprène peut s'écrire (C5H8) n avec n voisin de 10000. En d'autres termes, cet objet chimique mesure de l'ordre de 5 µm, ce qui justifie pleinement l'appellation de "macromolécule" proposée par l'allemand Hermann Staudinger dès 1926.
Si l'on veut résumer rapidement l'extraction nouvelle de matières naturelles ainsi que la synthèse des premiers produits innovants avant le début du XXe siècle, citons, chronologiquement mais pas nécessairement de manière exhaustive :
- dissolution du caoutchouc naturel à l'aide du naphta dès 1823 par l'écossais Mackintosh qui, conjuguée avec la vulcanisation découverte par l'américain Goodyear un matin de janvier 1840, permettra à l'anglais Sparow et au français Ader de doter les roues vélocipèdes de bandages de roues la même année dès 1868 ;
- mise au point en 1845 des premières bandes adhésives par l'américain Day ;
- en 1846, l'allemand Schönbein découvre précocement la nitrocellulose mais celle-là trouvera une utilisation tardive l'industrie de la chaussure vers 1900 ;
- en 1872, l'allemand Bayer étudie les réactions du formol sur les phénols ; mais le formol est un produit de laboratoire qui ne permet pas d'entrevoir une application industrielle rapide. Elle ne viendra qu'en 1912 dans la fabrication du contreplaqué entreprise par l'américain, d'origine belge, Baekeland ;
- en 1897, l'allemand Spitteler entreprend l'extraction de la caséine qui est la substance protidique constituant la majeure partie des albumines du lait et qui coagule sous l'action des acides. La caséine est également obtenue par caillage du lait sous l'action de ferments, le plus connu étant le labferment, qui est l'élément actif de la présure, obtenu par macération et raclage de la caillette du jeune veau. La colle de caséine fut adoptée immédiatement par l'aéronautique naissante pour le collage du bois (figure 18) et elles furent exclusivement utilisées par les allemands durant la guerre 1914-1918, pour le collage de toutes les pièces en bois des aéroplanes et des dirigeables.

Les colles de caséine, bien que donnant entière satisfaction seront rapidement détrônées par les colles d'urée formol dans les années 1925-1930, modernité oblige !
Le collage au XXe siècle
Avant toute chose signalons l'extraction de la gélatine en 1900 qui va trouver une application immédiate dans le collage du papier et de carton pour les travaux de reliure. En 1910, l'industrie du contreplaqué s'empare du silicate de soude, quant à la décennie suivante, c'est l'acétate de vinyle qui est envisagé pour le collage, mais sans application précise immédiate
C'est un peu avant la première guerre mondiale qu'apparaissent les colles fabriquées industriellement à partir du caoutchouc naturel. On les appelle sans faire preuve de beaucoup d'imagination "dissolutions". Elles trouvent leur application directe dans la confection des pneumatiques, en ce que l'on nomme aujourd'hui le collage par contact (figure 19). À cette époque, le pneumatique est fabriqué par simple application les unes sur les autres de plaques ou de bandes de caoutchouc enduites de "dissolution". Ces dissolutions sont encore actuellement utilisées pour réparer les chambres à air percées d'engins divers, à l'aide d'une rustine® prédécoupée de dimension adaptée.
Remarquons qu'il a fallu un temps certain, presque 50 ans, entre la première réaction de vulcanisation réalisée par Goodyear et son application directe à la fabrication d'un pneumatique par l'écossais Dunlop en 1888 qui réalisa tout d'abord un pneumatique à l'usage exclusif des bicyclettes, puis un modèle adapté aux véhicules hippomobiles et automobiles. Vétérinaire de profession, Dunlop fabriquait lui-même ses gants chirurgicaux en latex !

Durant l'entre-deux guerres les industriels restaient toujours tributaires plus ou moins directement des produits naturels, mais la révolution a lieu avec la fabrication des résines synthétiques et le développement de la chimie des macromolécules. En 1931, la société Du Pont de Nemours industrialise un caoutchouc "de luxe", le polychloroprène sous le nom déposé Néoprène®. Cette colle est bien connue des bricoleurs actuels, mais son inconvénient majeur est qu'elle colle à elle-même si bien qu'une fois étalée sur les surfaces à réunir, il faut attendre une temps certain l'évaporation partielle du solvant avant l'assemblage et le pressage.
Bien que les progrès scientifiques essentiels, qui ont permis la préparation des silicones et cela exclusivement en Europe entre 1823 et 1935 environ, c'est des Etats-Unis que les premières applications industrielles seront entrevues dès 1937, mais il faudra toutefois attendre 1941 pour que le chlorosilane soit breveté. Les polymères silicones présentent une excellente adhésion sur les matériaux à base de silice ou de silico-aluminates comme les verres. Les silicones constituent des produits adhésifs de choix pour les verres, soit pour les coller, soit pour assurer simplement le rôle de joints d'étanchéité et/ou de dilatation thermique. À titre d'exemple, les vitrages extérieurs de la Pyramide construite dans la cour du Louvre à Paris ont été jointoyés à l'aide de mastic silicone (figure 20). Le domaine du bâtiment, dont une grande partie des matériaux utilisés est à base de silice, constitue le secteur d'application privilégié des silicones. De plus, l'extrême facilité de mise en œuvre de ces matériaux permet l'ajout de résine et de silane pour faciliter le collage des matériaux présentant une importante énergie de surface, tels que les métaux et les matières plastiques à caractère polaire.
Le collage métal-métal apparaît dans les années 1940 à la fois aux USA, en Grande-Bretagne et en Allemagne, à la suite d'études entreprises sur le collage caoutchouc-métal, caoutchouc-bois et la mise au point des polyuréthanes et des polyisocyanates. La liaison métal - colle - métal était si résistante que l'on osait faire travailler mécaniquement ces assemblages et les utiliser dans la construction des avions (mais il arrivera par la suite que l'on déchante quelque peu) ; la notion de colle structurale était née, il s'agissait là d'une révolution extraordinaire. Actuellement, les pare-brise et vitres arrière des automobiles sont collés, non pas pour de simples raisons d'ordre esthétique, mais principalement pour participer à la résistance mécanique de l'habitacle du véhicule et améliorer concomitamment la sûreté des passagers. Ensuite, les résines époxydes voient le jour, permettant des collages sous faible pression ("PSA", sigle anglo-saxon pour désigner les Adhésifs Sensibles à la Pression).

Après la deuxième guerre mondiale, d'un art ou d'une technique, avec divers tours de main astucieux plus ou moins compliqués, le collage empirique devient une science, c'est-à-dire que commence l'étude des mécanismes du collage, du vieillissement et de la durée de vie des assemblages collés, divers modèles et théories seront proposés (ancrage mécanique, mouillage, modèle de l'interphase, effet électrostatique, modèle chimique, auto-adhésion, facteur dissipatif, effet ventouse, etc.). Bien évidemment, la compréhension commençante des mécanismes de l'adhésion fait que des produits nouveaux voient naturellement le jour, comme les cyanoacrylates, qui ne sont opérantes que sur des surfaces non acides, en présence d'humidité. Elles exige également un jeu faible entre les pièces à assembler.
De part sa rapidité d'exécution, un collage fort ancien connaît à nouveau une vogue extraordinaire comme le "Hot Melt" ou collage par fusion déjà signalé parce qu'utilisé par les égyptiens, en particulier avec les copolymères acétate de vinyle-éthylène (EVA), qui intéressent la reliure des livres, les chaussures, le contre-plaqué, les assemblages polymère-métal. Dans les grandes surfaces sont vendus des pistolets chauffants et des bâtons d'EVA qui satisfont parfaitement les besoins du bricoleur.
Pour le blocage des vis, la fixation et l'étanchéité, apparaissent les colles anaérobies, mises au point par l'américain Krieble en 1953, fondateur de la société Loctite®. La polymérisation de ces colles est catalysée par la présence de métaux en absence d'air. Elles trouvent leur utilisation en mécanique comme frein de filets des systèmes vis-écrou, susceptibles de se desserrer sous les effets de vibrations intempestives.
Quant à la décennie suivante, les années 1960, c'est au tour du développement des polyimides qui, en plus d'un bon comportement aux contraintes mécaniques, résistent à des températures de plus de 350 °C. Les colles conçues pour maintenir les tuiles sur les boucliers thermiques des sondes et navettes spatiales résistent à de bien plus hautes températures encore. Pour résoudre des problèmes de fixation de prothèses en chirurgie orthopédique et en orthodontie, des colles biocompatibles ont également été mises point.
Les principaux mécanismes du collage
Si l'on souhaite quelque peu respecter l'ordre chronologique, l'un des premiers modèles utilisés [10], dès 1926 par Mac Bain et Hopkins, pour rendre compte de l'adhésion durable entre deux solides est :
L'ancrage mécanique
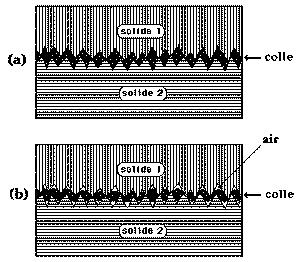
La pénétration par capillarité de la colle liquide dans les pores et entre les aspérités des surfaces en contact et la solidification ultérieure de cette colle crée un ancrage mécanique (figure 21). L'augmentation de la surface réelle de contact (a) et l'accroissement concomitant du nombre de liaisons inter-faciales permettent d'expliquer la forte adhésion constatée et l'accroissement de la résistance au cisaillement du joint dans le plan de l'interface. Encore faut-il, comme on le sait aujourd'hui, que la colle "mouille" le plus parfaitement possible les surfaces à réunir. Il ne faut surtout pas piéger des bulles d'air (b), qui sont autant de points de départ de fissures capables de ruiner l'assemblage collé.
Le mouillage
Les colles étant liquides, ou susceptibles de le devenir par chauffage, il n'est pas surprenant de prendre en compte la théorie du mouillage (figure 22). Au contact de la surface plane et lisse d'un solide, une goutte d'un liquide quelconque mouille plus ou moins la surface, c'est-à-dire qu'elle s'étale naturellement plus ou moins. La configuration adoptée par la goutte, définie par l'angle de contact θ, minimise l'énergie du système et rend parfaitement compte des interactions entre le liquide et le substrat solide. L'équation écrite par Thomas Young en 1805, prenant en compte les énergies inter-faciales entre le solide et le liquide γSL, le liquide et sa vapeur γLV et le solide avec la vapeur du liquide γSL (γSV = γSL + γLVcosθ) traduit cet équilibre. Un mouillage parfait correspond à θ = 0, comme on peut l'observer dans le cas de l'eau pure sur la surface propre d'une plaque de verre. On l'aura compris, la condition d'obtention d'une bonne adhésion dépend du mouillage au contact "colle liquide - surface solide" lors de la formation de l'assemblage. Il a été montré que pour qu'une goutte s'étale de manière satisfaisante (θ petit) il faut que sa tension superficielle soit inférieure à l'énergie superficielle du solide qu'elle recouvre.
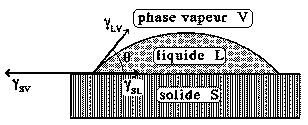
Ainsi, il convient de remarquer que si un solide A peut mouiller, à l'état fondu, un solide B, le solide B, à l'état fondu ne mouille pas le solide A. C'est précisément ce que l'on observe lorsque l'on souhaite réaliser un assemblage entre une résine époxyde et le polyéthylène. Le polyéthylène fondu déposé sur une surface de résine époxy fournit une bonne adhésion après refroidissement. En revanche, la polymérisation d'une résine époxyde (γB = 44 mJ.m-2) sur une surface de polyéthylène (γA = 31 mJ.m-2) ne donne pas d'adhésion.
Le modèle électrostatique
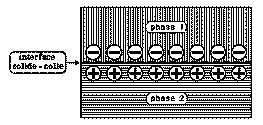
Pour des matériaux de natures différentes, chronologiquement il convient d'évoquer le modèle électrostatique (figure 23) qui fut développé à l'initiative de Deryagin et Krotova, de l'École russe à partir de 1948, suite à l'observation d'émissions d'électrons rapides lorsqu'un contact est rompu sous vide. Cette théorie est basée sur le transfert de charges électriques lors du contact, processus qui conduit en général à la formation d'une double couche électrique à l'interface, laquelle peut être assimilée aux deux plaques d'un condensateur plan. l'énergie de séparation est égale à (1/2)ρ2h/ε, où ρ représente la densité superficielle de charge, h la distance entre les deux plans du condensateur et ε la constante diélectrique du milieu. La distance h est calculée par la loi de Paschen qui lie le potentiel de décharge électrique à la pression du gaz dans lequel se produit cette décharge. Ce modèle, basé sur les attractions électrostatiques, n'est applicable qu'à des solides incompatibles, comme les assemblages verre-polymère et métal-polymère. De plus, ce phénomène est-il responsable de l'adhésion constatée ou en est-il la conséquence du fait de l'intimité du contact que l'on réalise ?
L'interphase
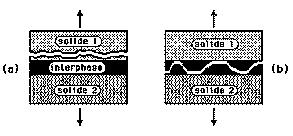
Dans les années 1960, l'américain Bikerman émit l'hypothèse de la formation au voisinage de l'interface d'une zone, l'interphase, caractérisée par une cohésion différente de celles des deux matériaux en présence (figure 24). Dans le cas d'une forte cohésion (a), une rupture ne peut se produire qu'à l'intérieur de l'un des solides. Au contraire, une rupture dans la zone de transition interfaciale traduit une faible cohésion (b). Ce modèle d'interphase, à forte ou faible cohésion, n'a pas la prétention d'expliquer les mécanismes de l'adhésion, par contre, il propose d'attirer une attention tout à fait particulière sur la nature de l'interface, après rupture, lorsque l'on affine l'échelle d'observation, du micromètre au nanomètre, c'est-à-dire à la dimension de la molécule.
La liaison chimique
Si l'interface est le siège de réactions chimiques créant des liaisons robustes de types covalent ou ionique conférant à l'assemblage une grande résistance à la rupture, comme c'est le cas avec les primaires d'adhésion, tels que les silanes, on parle de modèle chimique. À titre d'exemple dans le domaine de l'automobile : dans les pneumatiques, les fils d'acier constitutifs de la carcasse et de la bande de roulement du pneumatique, sont recouverts de laiton. L'adhésion de ces câbles au caoutchouc résulte de la formation de sulfures et de polysulfures de cuivre, à partir du cuivre constitutif du laiton et du soufre utilisé pour la vulcanisation du caoutchouc (figure 25).

L'inter-diffusion
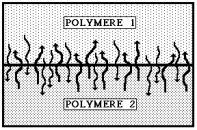
Un modèle voisin du précédent par l'aspect chimique qu'il représente est le modèle de l'inter-diffusion (figure 26), qui intéresse essentiellement les assemblages polymère - polymère de même ou de différentes natures chimiques (l'un étant constitutif du substrat et l'autre de la colle), pour lesquels les chaînes macromoléculaires des deux matériaux en contact diffusent à travers l'interface et réalisent la cohésion de l'ensemble. Cette théorie, proposée par Voyustkii de l'École russe dans les années 1950, permet d'expliquer les phénomènes de cicatrisation observés avec les polymères fracturés ou fissurés. Les mécanismes fondamentaux de diffusion des macromolécules ont été décrits et analysés finement par Pierre-Gilles de Gennes en utilisant le concept de reptation, le temps de relaxation associé à la reptation d'une chaîne variant comme le cube de la masse moléculaire du polymère. La distance de pénétration d'une macromolécule appartenant à une phase dans l'autre phase est proportionnelle à e-E'/R·Tt1/2 où E' est l'énergie d'activation de la diffusion et t le temps (la variation en racine carrée du temps est évidente pour la diffusion). Une question reste actuellement en suspens : l'inter-diffusion crée-t-elle l'adhésion ou se produit-elle à la suite d'une adhésion préalable ? C'est la même question que pour la théorie de l'électrostatique.
La dissipation rhéologique
Un dernier modèle en date, concerne plutôt l'adhérence que l'adhésion dans le sens où il fait intervenir la notion de rupture de l'assemblage collé plutôt que celle de sa cohésion, il s'agit du modèle du facteur dissipatif. Lorsqu'une fissure se propage dans un matériau ou à l'interface d'un joint adhésif, le bilan des énergies mises en jeu peut être écrit sous une forme très simple. L'énergie de rupture G est égale au produit de l'énergie de surface γ par le facteur Φ, sans dimension, de dissipation irréversible en volume, lequel résulte essentiellement des pertes visqueuses pour les adhésifs. Ce modèle permet de comprendre pourquoi il faut dépenser, pour rompre un assemblage collé, une énergie énorme, qui peut être 1000 fois, voire 10000 fois supérieure à l'énergie correspondant aux forces attractives entre les molécules.
Un mécanisme, qui amplifie l'effet du facteur dissipatif vu précédemment, a été récemment mis en évidence, en 1999, par les français Gay et Leibler [11], qui consiste en un effet ventouse créant de minuscules bulles d'air au sein de l'adhésif, qui se transforment en des cavités de plus en plus grosses, séparées d'abord par des parois puis par des filaments appelés également fibrilles qui se rompent à effort constant (figure 27). Ce phénomène, qui consomme de l'énergie, peut être aisément visualisé avec du chewing-gum® préalablement mâché maintenu entre le pouce et l'index.
A partir des modèles et théories précédemment décrits, il est possible de dégager quelques règles simples à suivre afin de réaliser un bon collage. En tout premier lieu, l'adhésif doit être appliqué sous forme liquide, sa viscosité étant la plus faible possible, il doit mouiller et nettoyer le substrat. L'adhésion peut être améliorée en augmentant la rugosité de la surface, ce qui facilite l'ancrage mécanique. Ensuite, on provoque la solidification de la colle qui doit devenir la plus visqueuse possible.
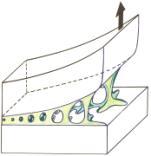
Avantages et incovénients du collage
Le collage trouve des applications dans tous les domaines industriels, il est devenu une technique d'assemblage au même titre que le triangle classique rivetage - vissage - soudage. Il est vrai que le collage présente un très grand nombre d'avantages tant du point de vue de sa mise en œuvre que de celui de la qualité des assemblages réalisés ; en contrepartie, certaines précautions d'emploi doivent être observées, et la conception des pièces doit nécessairement prendre en compte ce procédé particulier d'assemblage.
Pour ce qui concerne les avantages, signalons en tout premier lieu, l'augmentation de la cadence de fabrication, par suite, d'une part, d'un moindre besoin de pièces à assembler (vis, rondelles, écrous, rivets, etc.) et d'autre part, de la rapidité de l'application du produit adhésif qui, de plus, est souvent automatisée. Le collage est parfaitement adapté à l'assemblage des matériaux différents, des matériaux fragiles et des matériaux minces : la colle forme un joint continu entre les solides au travers duquel se transmettent parfaitement les efforts mécaniques (figure 28). La résistance mécanique est supérieure à celle produite par le rivetage, à moins de placer un rivet tous les millimètres, mais cela aurait pour conséquence immédiate de provoquer un déchirement.
Plus particulièrement pour les matériaux métalliques, par rapport au soudage à haute température, le collage permet de conserver les caractéristiques obtenues par les traitements thermiques réalisés si bien qu'il n'entraîne pas de modification des dimensions des pièces et, au besoin, permet même de rattraper les dépassements de tolérances d'usinage. Le gain de poids par rapport aux techniques mécaniques énumérées précédemment intéresse particulièrement les applications destinées à l'aéronautique ainsi qu'à l'astronautique. Le fait d'obtenir un joint de colle continu supposé étanche évite les couples galvaniques lors de l'assemblage de métaux différents.
N'oublions pas non plus l'amélioration de l'aspect esthétique apportée par le collage, les surfaces extérieures des assemblages pouvant être parfaitement lisses, ce qui ne peut qu'améliorer la résistance à l'air en aéronautique, par exemple. Autre avantage : les films de colle peuvent à volonté absorber ou amplifier les vibrations, et apportent une isolation à la fois électrique, thermique et phonique.
Venons-en maintenant aux inconvénients. Ils sont nombreux et importants. L'opération d'assemblage implique la résolution incontournable de trois problèmes :
- à partir du moment où l'on a décidé de coller, c'est-à-dire dès la conception au bureau d'études, il faut choisir des formes et des dimensions adéquates pour les surfaces des solides à réunir en évitant, en particulier, toute concentration de contraintes si la structure est destinée à en supporter ;
- il faut préparer les surfaces des solides à coller, autrement dit, il faut appliquer aux surfaces des traitements mécaniques et physico-chimiques, opérations en général coûteuses mais indispensables pour éliminer les impuretés, augmenter l'énergie de la surface, accroître l'accrochage mécanique et favoriser le mouillage de la colle ;
- il faut choisir le produit adhésif en fonction de nombreux critères liés à la nature même des solides, aux conditions auxquelles sera soumis l'assemblage, à la forme liquide ou solide à bas point de fusion de l'adhésif, à ses caractéristiques de mouillabilité, à sa facilité d'application sur les surfaces ainsi qu'aux conditions et à la durée de la solidification, temps de séchage (durée de prise de la colle), par exemple.
Quant au joint adhésif ainsi réalisé, dont le démontage et le repositionnement des éléments sont deux opérations impossibles à envisager sur une chaîne de fabrication, on souhaite en contrôler la qualité, en mesurer la résistance mécanique et en estimer la durée de vie. Ces paramètres ne sont accessibles qu'à partir des résultats d'une étude préliminaire fine, et/ou d'expériences témoins sur éprouvettes conduites préalablement ou en parallèle pendant la fabrication, consistant à détruire le produit collé. Il existe aussi des contrôles non destructifs, ils sont utilisés chaque fois que la forme, la structure et les dimensions de l'assemblage collé le permettent.
De plus, parmi toutes les colles existantes, certaines ne résistent pas aux contraintes mécaniques appliquées, à la chaleur, au froid ou encore aux chocs thermiques, d'autres présentent un mauvais comportement à certaines radiations, d'autres encore, techniquement parfaites pour l'utilisation envisagée, s'avèrent trop onéreuses compte tenu de la grande superficie à encoller, si bien qu'il n'existe pas de colle universelle alliant toutes les qualités à la fois. Savoir coller, c'est trouver le bon compromis.
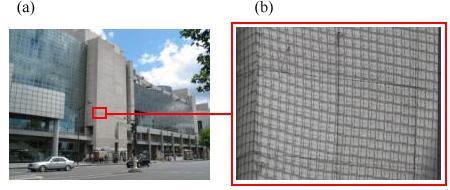
Un paramètre souvent omis par les architectes et les maîtres d'ouvrages est la capacité de la colle à accommoder les écarts de dilatation thermique des matériaux assemblés. C'est pourquoi, la façade de certains édifices, comme l'Opéra Bastille à Paris (figure 29), sont couvert par un filet de protection afin d'éviter que les dalles en marbre du parement, en se décollant, tombent sur les passants.
Les différents types de colle
Les adhésifs ou colles utilisés pour réaliser l'assemblage d'éléments et assurer l'étanchéité peuvent être classés, selon la nature chimique de leur principal composant, celle des couples de matériaux à coller entre eux ou encore suivant les mécanismes ou techniques mis en jeu.
Pour l'utilisateur, qu'il soit industriel, artisan ou simple bricoleur, le choix est essentiellement guidé par la nature des éléments à coller et la simplicité d'utilisation de l'adhésif adéquat, selon les indications plus ou moins claires, fournies par le fabricant. En d'autres termes, il existe des colles ne nécessitant pas ou peu de préparation alors que d'autres ne sont opérantes qu'à la suite d'un protocole expérimental particulier ou/et d'une réaction chimique dont la réalisation demande des précautions certaines. Il semble donc judicieux, comme l'a proposé en 1992 Eduardo Schindel-Bidinelli, de classer les colles suivant deux catégories : celles qui ne font appel qu'à des mécanismes physiques et celles pour lesquelles une réaction chimique est nécessaire. Pour ce qui concerne les processus physiques, citons à titre d'exemples les trois principaux utilisés : l'évaporation d'un solvant, aqueux ou organique, l'application d'une force sur l'assemblage dans le but d'augmenter l'intimité du contact entre les deux surfaces à réunir et l'échauffement de l'adhésif en vue de le ramollir ou de le fondre, suivi d'un refroidissement lent ou rapide, selon le cas, afin de provoquer son durcissement.
Les colles à mécanismes physiques
Parmi les colles faisant intervenir un processus physique, les plus simples à utiliser, signalons celles dont le principe actif est en solution ou en suspension dans l'eau, le collage s'effectuant par évaporation. Elles sont bien connues du grand public essentiellement parce qu'elles ne demandent aucune préparation avant emploi et qu'elles conviennent pour un grand nombre de matériaux usuels, même poreux. Ces colles sont aussi bien d'origines naturelles (minérales, végétales et animales) que synthétiques. Parmi les colles naturelles, signalons les adhésifs à base de silicate de soude, dextrines et amidons pour les industries du papier et du carton, à base de ciment et de bitume pour le collage des carrelages, la gomme arabique pour l'enduction des timbres et étiquettes, les produits à base de colophanes pour le collage des revêtements de sols, les colles à base de caséine du lait pour la fabrication des structures en bois lamellé-collé.
Les colles d'origine synthétique en milieu aqueux sont des adhésifs phénoliques, utilisés dans l'industrie du bois pour le contreplaqué et le lamellé-collé, il y a également les colles d'urée-formol pour le plaquage en menuiserie et ébénisterie, les alcools polyvinyliques et l'acétate de polyvinyle pour le bois, le carton et le papier, sans oublier le polyvinyle-pirrolidone pour la colle dite "de bureau".
Tout aussi simples à utiliser sont les adhésifs synthétiques à solvants organiques, le collage s'effectuant par évaporation du solvant. Ces colles sont à base d'élastomères, comme les polychloroprènes (Néoprène®) et les polyuréthanes pour le collage des textiles et pour l'industrie de la chaussure, à base vinylique pour le bricolage (éthers polyvinyliques). Font aussi partie de cette catégorie, les aminoplastes et phénoplastes pour le collage des stratifiés, les polyuréthanes utilisés dans les industries de l'automobile et de l'aéronautique, les néoprène-phénolique, nitrile-phénolique et vinyle-phénolique pour les collages structuraux. Comme cela a déjà été signalé, le qualificatif "structural" signifie que le rôle principal de l'adhésif est de créer une liaison mécanique forte et rigide entre les éléments déformables constituant l'assemblage ; l'adhésif employé fait partie intégrante de l'ensemble de la structure et les assemblages ainsi réalisés peuvent supporter des contraintes aussi importantes que les systèmes mécano-soudés (figure 30).
De nouveaux adhésifs structuraux, dits renforcés, apparaissent sur le marché, le renforcement consistant en l'incorporation, dans une colle réputée rigide et cassante telle que peut l'être une résine acrylique, de micro-bulles de caoutchouc, lesquelles augmentent la résistance à la rupture.
Doivent être également mentionnés les produits auto-adhésifs conditionnés ordinairement sous forme de ruban ou de plaque (Scotch®, Tesa®). Ils sont fabriqués principalement à base de caoutchoucs naturel et synthétiques et de polyacrylates. Ils agissent par une légère pression sur l'assemblage (étiquettes et plaquettes autocollantes, par exemple) et conviennent aux matériaux les plus divers, y compris les métaux et matières plastiques, la condition de porosité n'étant pas impérative.
Pour le cas particulier du collage entre elles des matières thermoplastiques (lesquelles ramollissent et fondent par chauffage et de manière réversible se solidifient et se rigidifient par refroidissement), on utilise le mécanisme de la diffusion en employant des produits à base de solvant organique, comme le Tangit® pour le collage d'éléments en chlorure de polyvinyle, par exemple. Les objets en plexiglas® sont collés selon le même principe, à l'aide d'une solution de Plexiglas®, l'utilisation simultanée d'ultrasons accélérant le processus de diffusion.
Il est difficile de distinguer fondamentalement les adhésifs dits "à solvant", examinés précédemment, des adhésifs dits "de contact" du fait même que l'assemblage est réalisé après évaporation complète du solvant organique. La spécification "de contact" est introduite pour rendre compte que le collage nécessite l'application d'une légère pression. Les colles entrant dans cette catégorie sont à base de polyuréthane, de copolymères styrène-butadiène ou encore de polychlorobutadiène et sont principalement utilisées pour réaliser des assemblages composites cuir - textile, cuir - caoutchouc, tels qu'on peut les rencontrer dans l'industrie de la chaussure, et matière plastique - béton et textile - béton pour ce qui concerne la pose des revêtements de sols ou muraux. De manière générale, les surfaces à réunir sont, avant pression l'une contre l'autre, préalablement enduites et séchées.
Un type complètement différent de colles, dites thermo-fusibles, peuvent être utilisées concurremment avec celles évoquées précédemment. Ces adhésifs, qui sont sans solvant, sont appliqués fondus et c'est la couche refroidie, plus ou moins souple, qui constitue le système d'assemblage. Ces produits, à base de copolymères styrène-butadiène ou éthylène-acétate de vinyle (EVA) ou encore à base de polyester, interviennent dans un grand nombre de domaines industriels tels que : l'automobile, l'ameublement, l'électronique, l'habillement et la reliure, par exemple.
Les colles à mécanismes chimiques
Les colles modernes sont des polymères artificiels, c'est-à-dire des macromolécules dont la synthèse consiste à enchaîner les unes aux autres des molécules de petites dimensions en pratiquant des réactions chimiques spécifiques. Celles-ci sont des polymérisations ou copolymérisations radicalaires (par ouverture de liaisons multiples sous l'effet d'interventions extérieures, telles que chauffage et rayonnements ultraviolets, par exemple), des polycondensations (telles que des estérifications, avec élimination d'eau et des substances légères acides ou alcooliques) et des polyadditions, utilisées moins fréquemment que les précédentes, qui consistent en l'addition répétée d'un élément monomère porteur d'un radical fonctionnel.
Parmi les adhésifs qui deviennent opérants sous l'effet d'une action extérieure, il existe les colles qui durcissent grâce à l'humidité de l'air ambiant. On distingue celles à base de polyuréthane ou de polysiloxane (colles silicone) et celles à base d'esters de l'acide cyanoacrylique. Elles conviennent pour coller les métaux, les matières plastiques, les élastomères, et sont plutôt adaptées aux surfaces de faible dimension pour lesquelles la pénétration de l'humidité est favorisée. À température ambiante, l'humidité relative optimale est de l'ordre de 65 %.
Il existe des produits, polyesters et polyuréthanes comportant des doubles liaisons ainsi que des acrylates, qui sont radio-sensibles, c'est-à-dire que l'exposition à une radiation ultraviolette provoque leur durcissement, d'où l'idée de les utiliser pour réaliser des colles. Celles-ci sont parfaitement adaptées à l'assemblage des verres, céramiques, métaux et matières plastiques, la seule condition étant que l'un des éléments à réunir soit transparent à la longueur d'onde (100-380 nm) de la radiation ultraviolette, cet élément étant, par conséquent, nécessairement de très faible épaisseur. Ces résines photo-polymérisables sont très largement utilisées en Odontologie, par exemple.
Un dernier type de colles apparu dans les années 1950-55, dont on peut dire également que le durcissement est dû à une action extérieure, est représenté par les adhésifs anaérobies. Ces produits, à base de di-acrylate, durcissent par effet catalytique au contact des métaux en absence d'oxygène ou d'air et conviennent donc parfaitement au collage des matériaux métalliques. On les utilise très souvent à des fins de blocages : blocage des emmanchements cylindriques axe-tourillon lisses et blocage des vis, la colle liquide comble le jeu entre les pièces ou l'espace libre entre les filets des vis et écrous et ceux des vis et trous taraudés (Loctite®, par exemple). Après durcissement, non seulement les pièces sont bloquées par empêchement de tout glissement relatif, mais aussi les assemblages sont rendus étanches, ce qui présente l'avantage certain de supprimer ou de diminuer très fortement le risque de corrosion. Le joint de colle peut également jouer éventuellement un rôle d'amortissement des vibrations.
Il existe un très large éventail de colles nécessitant le mélange de deux composants, appelés communément résine et durcisseur, afin de produire la réaction chimique. Les colles "à réaction chimique" sont relativement simples à utiliser. Il suffit de mélanger la résine et le durcisseur dans les proportions prescrites par le fabricant, en évitant toute inclusion de bulles d'air, lesquelles affaibliraient considérablement la résistance du joint adhésif formé. Relèvent de ce premier type de colles, les résines époxydes et acryliques, les polyesters qui conviennent à tous les matériaux utilisés dans les industries aéronautiques, mécaniques, métallurgiques, tels que alliages métalliques, céramiques, élastomères, matières plastiques, roches, verres, etc. À titre d'exemple, pour les époxydes, qui sont des adhésifs thermodurcissables, la résine de base est le Diglycidyléther du Biphénol A (DGEBA) dont la polymérisation est assurée grâce à des amines ou diamines, lesquelles jouent ainsi le rôle de durcisseur. Afin de conférer à ces colles des propriétés particulières, plusieurs ingrédients viennent compléter la formulation tels que talc, kaolin et caoutchouc. Les Araldite® entrent dans cette catégorie de produits adhésifs.
Les mêmes matériaux que ceux énumérés précédemment peuvent être collés avec des produits, de conception astucieuse, déclarés mono-composants parce que l'adhésif, une résine époxyde par exemple, contient à l'état latent le durcisseur (en général, il s'agit du dicyan-diamide, DiCy). La réaction de polymérisation est déclenchée par élévation de la température, les éléments à assembler étant maintenus sous une légère pression. Bien évidemment la résistance du joint adhésif dépend fortement des conditions opératoires recommandées par le fabricant concernant la température et la durée du chauffage. On trouve actuellement dans le commerce des "pâtes à coller" (Bostik®, par exemple), qui sont capable de fixer sur tous supports n'importe quel objet. À y bien regarder, ces pâtes, qui se présentent sous forme de bâtons, contiennent deux composants malléables positionnés de manière concentrique, qu'il faut malaxer pendant quelques minutes pour homogénéiser le mélange et provoquer le durcissement futur de ces colles appréciées des bricoleurs.
Un dernier type de colles à réaction chimique, à base de résines phénol-formol, résines époxydes et phénoliques, demande le plus grand soin de préparation puisqu'on dispose de deux composants séparés à mélanger et que les éléments de la structure à assembler doivent être chauffés sous pression. Il faut donc impérativement réaliser le mélange dans les conditions qu'impose rigoureusement le fabricant afin de créer un joint adhésif parfait, résistant et durable, ce qui nécessite par conséquent la maîtrise complète des proportions des composants, de la température des pièces en contact, de la force de pression, celle-là étant appliquée pendant la durée préconisée.
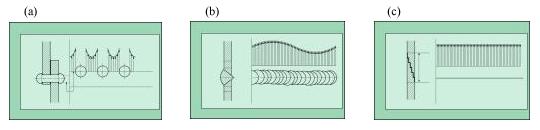
Les différents types de collage
Les quatre principaux types de collage sont représentés schématiquement dans la figure 31.

1. - le collage en bout (1 et 4 dans la figure 31), ne supportent pas d'importants efforts mécaniques. Mais, utilisé naguère par le "raccommodeur de faïences et de porcelaines", qui réparait pour quelques sous les vaisselles brisées, est toujours pratiqué de nos jours par les spécialistes de la restauration d'objets archéologiques, généralement des poteries à reconstituer à partir de fragments plus ou moins gros ;
2. - le collage par recouvrement (2, 3, 5 et 11 dans la figure 31), est beaucoup plus résistant que le précédent et surtout le plus fréquemment utilisé, par exemple, pour réunir les deux parties d'une feuille de papier déchirée à l'aide d'un ruban adhésif, pour cacheter une enveloppe, appliquer un timbre autocollant, pour ressemeler une chaussure, pour fixer un revêtement mural, une moquette, pour assembler les divers éléments nécessaires à la réalisation d'un boîtier de montre (mêmes les plus coûteuses sont collées !), pour fixer un pare-brise et la vitre arrière sur une carrosserie, pour poser un pansement adhésif ou un tulle enduit en vue d'une épilation, , pour étiqueter une bouteille, pour fabriquer un bouchon de champagne, pour fixer les verrous orthodontiques en vue d'embellir une denture déformée par une trop longue période de suçage de pouce, par exemple, etc. ;
3. - le collage mixte, c'est-à-dire en bout et par recouvrement (6, 7, 8, 9 et 10 dans la figure 31), est utilisé pour confectionner des structures complexes, comme les skis constitués de matériaux divers, comme les charpentes en bois lamellé-collé. Une telle charpente, de forme sphérique, soutient actuellement le dôme de la salle de cinéma Imax en bordure de l'esplanade à Paris-La Défense (figure 32) ;

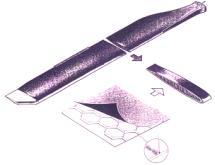
4 - le collage selon la structure "nids d'abeilles", lequel associe robustesse, élasticité et légèreté à l'assemblage collé, est utilisé dans la fabrication des pales d'hélicoptère (figure 33) et confère une grande fiabilité à ces organes vitaux. Actuellement, tous les éléments constitutifs de la structure d'un hélicoptère moderne sont collés.
Conclusion
A l'heure actuelle, les colles d'origines naturelles, végétale, animale et minérale, sont toujours utilisées mais en les combinant le plus souvent avec des adhésifs synthétiques. Il existe actuellement une bonne dizaine de milliers de formulations de colles ou adhésifs. En fait, à partir de quelques résines de base, c'est le jeu des mélanges et le nombre des additifs qui conduisent à un nombre aussi élevé. Pour obtenir une propriété donnée, on utilise une charge incorporée à la résine et cela fournit une nouvelle formulation. Beaucoup de produits sont en réalité équivalents mais commercialisés sous des appellations différentes. Le collage simplifie grandement notre vie de tous les jours, en utilisant par exemple, les étiquettes autocollantes et les divers rubans adhésifs qui permettent d'assembler par juxtaposition ou/et par superposition. Les bandes adhésives remplacent avantageusement les épingles, dites de sûreté, utilisées naguère pour les couches de bébés.
Toutefois, on peut regretter que malgré, ou du fait de, la quasi quotidienneté d'actes de collage divers, le langage populaire associe encore aux colles la notion de produits "bas de gamme" aux propriétés mécaniques médiocres et aux adhésifs ou résines, celle de composés technologiquement très élaborés possédant une capacité importante à résister à d'importantes contraintes, qu'elles soient mécaniques et/ou thermiques. En fait, aujourd'hui, on peut affirmer que l'on peut tout coller, mais pas avec n'importe quoi et surtout pas en s'y prenant n'importe comment.
Le problème actuel est ailleurs, c'est celui de la prédiction de la durée de vie des joints collés. L'expérimentation sur éprouvettes montre que les adhésifs structuraux, dans une atmosphère tempérée non trop humide, ne voient pas leurs caractéristiques évoluer en l'absence de fortes contraintes extérieures. Par contre, si la contrainte imposée excède environ 20% de la contrainte de rupture, la durée de vie du joint décroît exponentiellement avec l'intensité de la contrainte, quand cette dernière est imposée de manière permanente.
Le paramètre le plus important semble être l'humidité : un joint collé perd 90% de sa capacité à résister à la rupture lorsqu'il est saturé d'eau. Heureusement, d'une part l'imprégnation d'eau est réversible, c'est-à-dire que le joint, à moins d'une détérioration, retrouve ses propriétés initiales dès que le taux d'humidité revient à un taux normal et d'autre part, la diffusion de l'eau est très lente.
En conclusion, l'histoire n'est pas tout à fait finie : la prédiction de la durée de vie d'un joint collé n'est que pour demain !
Références
[1] K. Autumn, Y.A. Liang, S.T. Hsleh, W. Zesch, W.P. Chan, T.W. Kenny, R. Fearing, R.J. Full, «Adhesive force of a single gecko foot-hair» Nature, 405 (2000) 681-685.
[2] A.K. Geim, S.V. Dubonos, I.V. Grigorieva, K.S. Novoselov, A.A. Zukhov, S.Y. Shapovalm, «Microfabricated adhesive mimicking gecko foot-hair» Nature materials, (2003) 1-3.
[3] M. Regert, «Investigating the history of prehistoric glues through gas chromatography - mass spectroscopy» Journal of separation science , 27 (2004) 244-254.
[4] D. Stordeur, La main et l'outil, manches et emmanchements préhistoriques, Maison de l'Orient, diffusion de Boccard, Paris, 1987.
[5] P.Anderson, J.Chabot, «La première machine agricole et les lames cananéennes» Dossiers d'Archéologie , 290 (2004) 44-51.
[6] E.Boëda, J.Connan, D.Dessort, S.Muhesen, N.Mercier, H.Valladas, N.Tlsnérat, «Bitumen as a hafting material on middle paleolithic artefacts» Nature , 380 (1996) 336-337.
Pour en savoir plus
- J.Bost, Matières plastiques, Lavoisier, Paris, 1985.
- J.Cognard, Science et technologie du collage, PPUR, Lausanne, 2000.
- P.Couvrat, Le collage structural moderne, Lavoisier, Paris, 1992.
- M.Daumas, Histoire générale des techniques, Quadrige / PUF, Paris, 1996.
- J.D.Ferry, Viscoelastic properties of polymers, John Wiley & Sons, Inc., New York, 3e édition, 1980.
- P.G.de Gennes, Scaling concepts in polymer physics, Cornell University Press, Ithaca, New York, 2e édition, 1985.
- J.N.Israelachvili, Intermolecular and surface forces, Academic Press, 2e édition, Londres, 1997.
- J.J.Meynis de Paulin, Les colles et adhésifs, Éditions Guy Le Prat, Paris, 1974.
- E.H.Schindel-Bidinelli, Pratique du collage industriel, Lavoisier, Paris, 1992.
- M.Soutif, L'Asie, source de sciences et de techniques, PUG, Grenoble, 1995.