Cet article est consacré à la recherche de nouveaux principes actifs in silico (drug design in silico), en se limitant aux principes actifs constitués de petites molécules, à l'exclusion des vaccins et biomédicaments. Il fait suite à un premier volet présentant les bases de données disponibles en libre accès regroupant cibles thérapeutiques et petites molécules chimiques susceptibles d'interagir avec ces cibles. Ce second volet traite du criblage virtuel en ligne et des deux principales approches de celui-ci : celle qui utilise les propriétés des petites molécules chimiques - en anglais, ligand-based virtual screening (LBVS) - et celle qui utilise la structure tridimensionnelle de la cible thérapeutique - en anglais, structure-based virtual screening (SBVS).
Vers une recherche de petites molécules chimiques sur internet : le criblage virtuel en ligne
Criblage expérimental et criblage virtuel
Dans les années 1990, le processus de recherche de petites molécules chimiques a connu une mutation sans précédent. On assiste alors à l’adoption de techniques de criblage massif parallèles. Le criblage robotisé à haut débit change alors radicalement le quotidien des chercheurs de médicaments. On ne teste plus manuellement les petites molécules chimiques comme c’était le cas auparavant, mais ce sont des automates qui accomplissent cette tâche. Parallèlement, les robots de synthèse sont capables de produire des dizaines de milliers de composés chimiques chaque jour qui seront ensuite stockés dans une chimiothèque. Cependant, malgré ces révolutions technologiques, le processus de recherche d'un médicament chimique reste encore semé d’embûches.
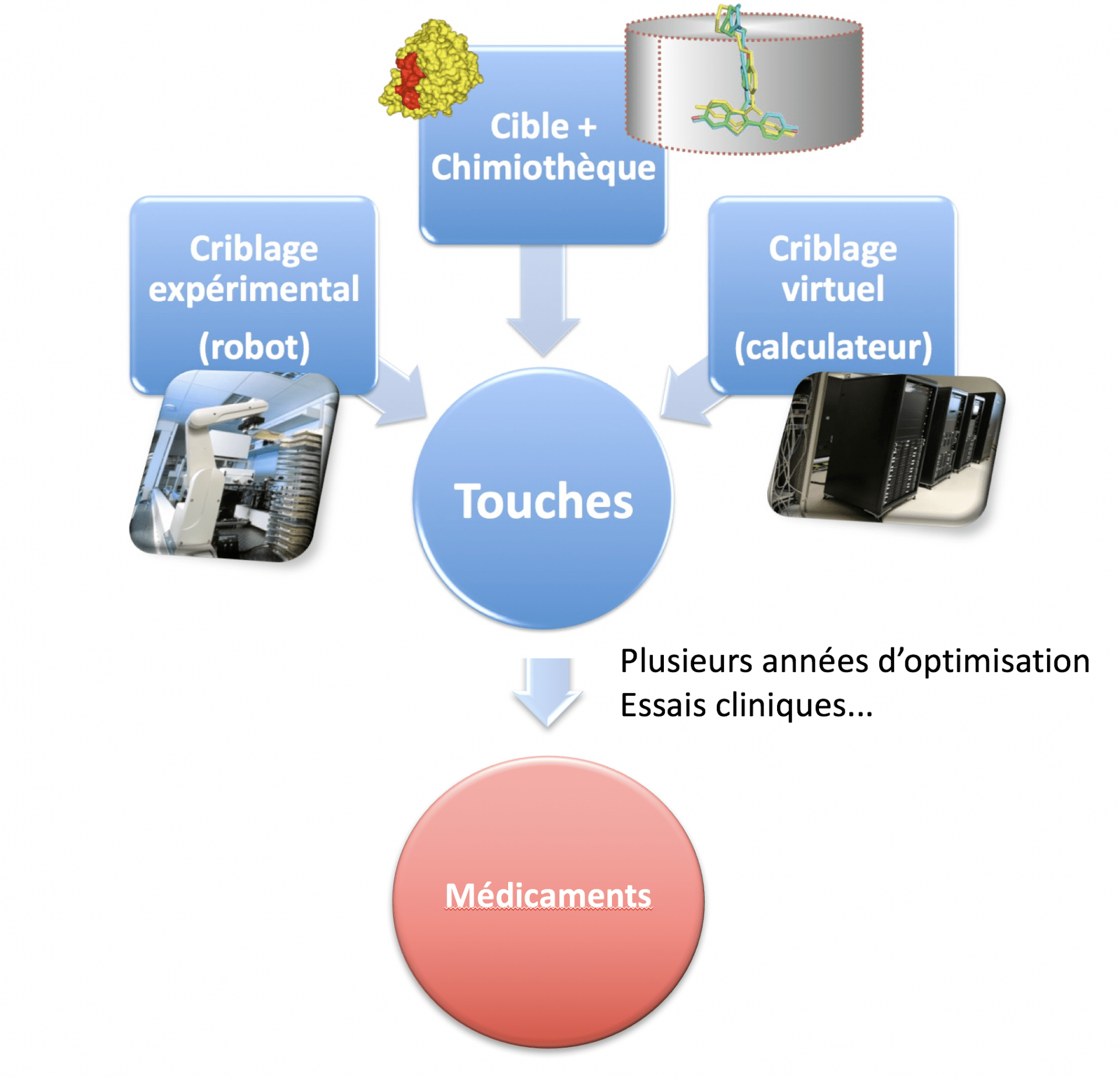
Le criblage expérimental robotisé et le criblage virtuel permettent d’évaluer des milliers ou des millions de petites molécules, stockées dans une chimiothèque (réelle ou virtuelle) et d’identifier des molécules « touches » actives sur une cible ou une cellule.
Le criblage expérimental, appelé « screening » en anglais (ou « high-throughput screening » ou HTS pour souligner le caractère haut-débit de l’approche), est une technique de référence pour la recherche de petites molécules agissants sur une cible (Fig. 1). Dans la pratique, il consiste à tester en parallèle au moyen d'un automate l'action de dizaines de milliers de petites molécules chimiques sur une cible1 (par exemple, une protéine) que l’on estime importante pour une pathologie donnée, dans le but d'identifier des touches (appelées « Hits » en anglais) qui vont agir sur la cible ou les cibles (appelées « Target(s) » en anglais). Avec la miniaturisation et l’automatisation des tests biologiques, l’automate peut évaluer 100 000 molécules par jour environ (voire plus), mais les coûts sont énormes. Pour visualiser les réactions issues de la mise en contact du produit chimique avec la cible, on utilise différentes méthodes basées sur des mesures d’absorbance, de luminescence, de fluorescence ou encore faisant appel aux techniques d’imagerie.
Les touches intéressantes issues du criblage sont alors ré-évaluées, dans des tests biologiques plus précis. Les plus prometteuses seront ensuite modifiées chimiquement afin d’augmenter leur affinité et leur sélectivité pour la cible. Conjointement, certaines de leurs propriétés d’absorption, de distribution, de métabolisme, d’excrétion et de toxicité (ADME-Tox) seront optimisées pour devenir compatibles avec une administration ultérieure chez l’homme. Mais même à ce stade, on est encore loin du médicament ! Le processus d’optimisation continuera encore pendant plusieurs années, le rapport efficacité/dose, la toxicité éventuelle, les effets secondaires et le choix de la formulation devront être étudiés, jusqu’à ce que le produit puisse entrer en phase clinique. Dans le cas contraire, si les molécules restent toxiques ou ont des effets indésirables détectables à ce stade ou si leur efficacité est insuffisante, en dépit d’efforts multiples, elles seront abandonnées, ce qui entraîne la frustration intellectuelle bien légitime du chercheur et des pertes financières considérables.
Le criblage expérimental traditionnel laisse toujours une part importante au hasard, en s’appuyant souvent sur la logique des grands nombres : la probabilité de trouver une petite molécule active est proportionnelle au nombre de produits chimiques testés. Pour améliorer les résultats il est nécessaire de s’affranchir en partie de cette vision purement aléatoire en utilisant notamment les techniques de criblage virtuel, les approches de bioinformatique structurale et de chémoinformatique associées à des études biophysiques.
Si plusieurs approches informatiques ont été développées ces dernières décennies pour assister et rationaliser le processus de recherche de molécules thérapeutiques, ce n’est qu’à partir des années 2000 que la chémoinformatique et le criblage virtuel deviennent vraiment opérationnels. De nos jours, ces protocoles sont incontournables dans un projet moderne de recherche de nouveaux médicaments chimiques. La chémoinformatique est au carrefour de multiples disciplines et ses deux applications principales sont : d’une part, l’organisation et la recherche de l’information chimique et, d’autre part, l’identification de corrélations entre la structure des petites molécules chimiques et leur « succès » ou activité dans les tests biologiques. Les concepts chémoinformatiques sont utilisés non seulement dans le criblage virtuel mais aussi dans les projets visant à l’anticipation des risques de toxicité et d’écotoxicité, dans la préparation des chimiothèques et dans d’autres domaines qui dépassent le cadre de la seule chimie médicinale.
Le criblage virtuel, dans sa version la plus simple, transpose in silico certaines idées du criblage expérimental avec cependant des différences notables (Fig. 1). Le criblage virtuel a en effet pour objectif principal de réduire le nombre de molécules à tester expérimentalement mais sert également à guider le chimiste dans la sélection de molécules aux propriétés physico-chimiques compatibles avec le développement d’un médicament. Il faut distinguer deux grandes stratégies de criblage virtuel qui peuvent être couplées dans certaines circonstances (Fig. 2) :
- celle qui utilise les propriétés structurales des petites molécules chimiques bioactives, on parle alors de criblage virtuel basé sur la structure des ligands1(« ligand-based virtual screening » ou LBVS) (Fig. 3),
- celle qui utilise la structure tridimensionnelle (3D) de la cible (« structure-based virtual screening» ou SBVS) (Fig. 4).
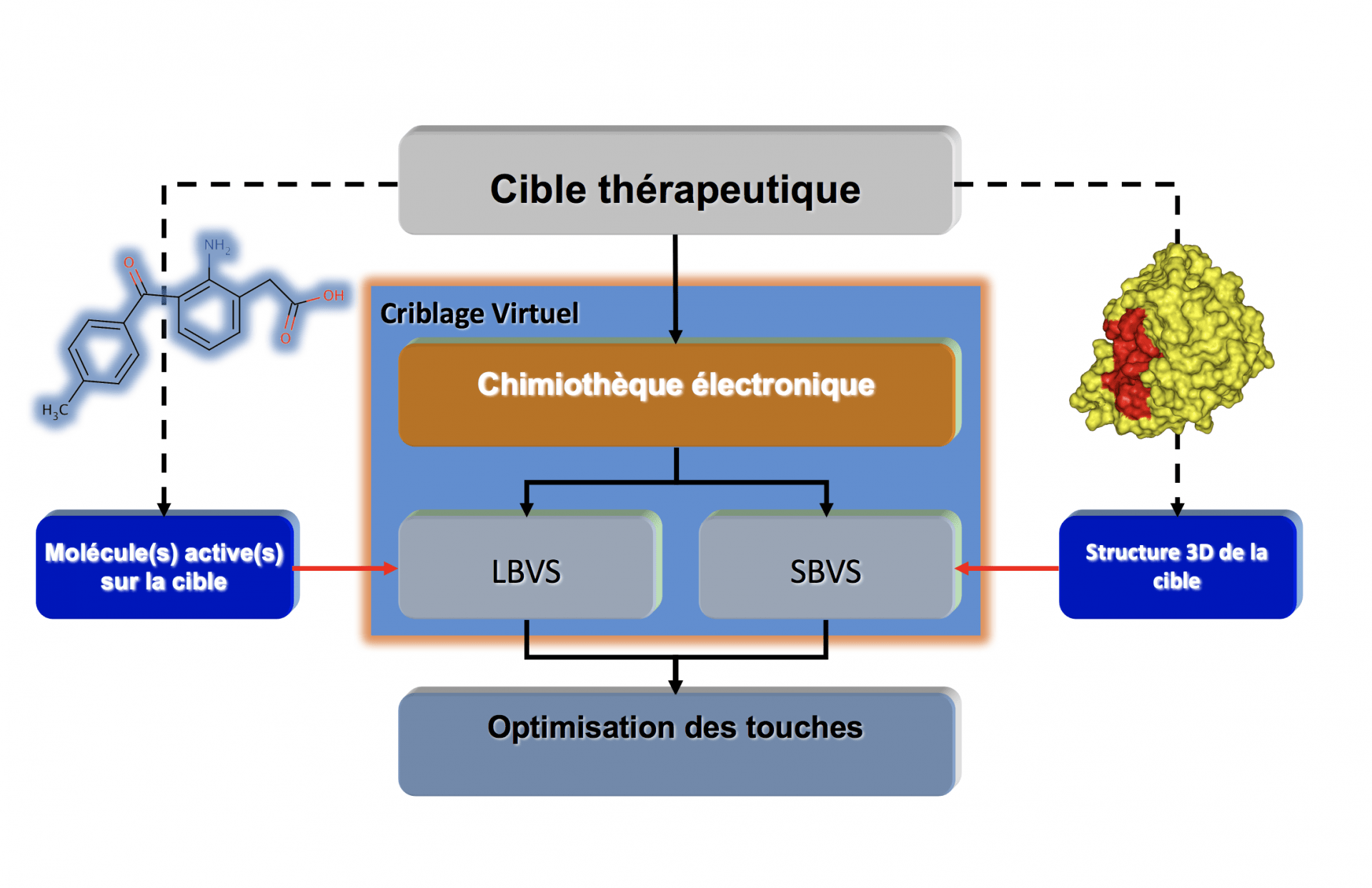
Dans les deux cas, une chimiothèque électronique pouvant contenir plusieurs millions de molécules sera généralement utilisée. Une petite liste de molécules (100 à 500) à tester expérimentalement sera générée.
Le criblage virtuel, la bioinformatique structurale, qui analyse les cibles et les sites de fixations des candidats médicaments, et la chémoinformatique révolutionnent certaines étapes-clés de la recherche de molécules thérapeutiques, car ces approches permettent de mieux préparer les chimiothèques, de réduire le nombre de produits à tester in vitro ou in vivo et facilitent l’optimisation des touches. Par exemple, à partir d’une chimiothèque électronique initiale contenant 1 million de petites molécules, le criblage virtuel permettra, en quelques heures de calculs (ou en quelques minutes de calcul sur un cluster puissant), de générer une liste d’environ 500 touches potentiellement actives à tester expérimentalement. Sur ces 500 molécules analysées en tubes à essai, il y aura immanquablement un nombre important de molécules inactives mais aussi une petite liste de molécules bioactives.
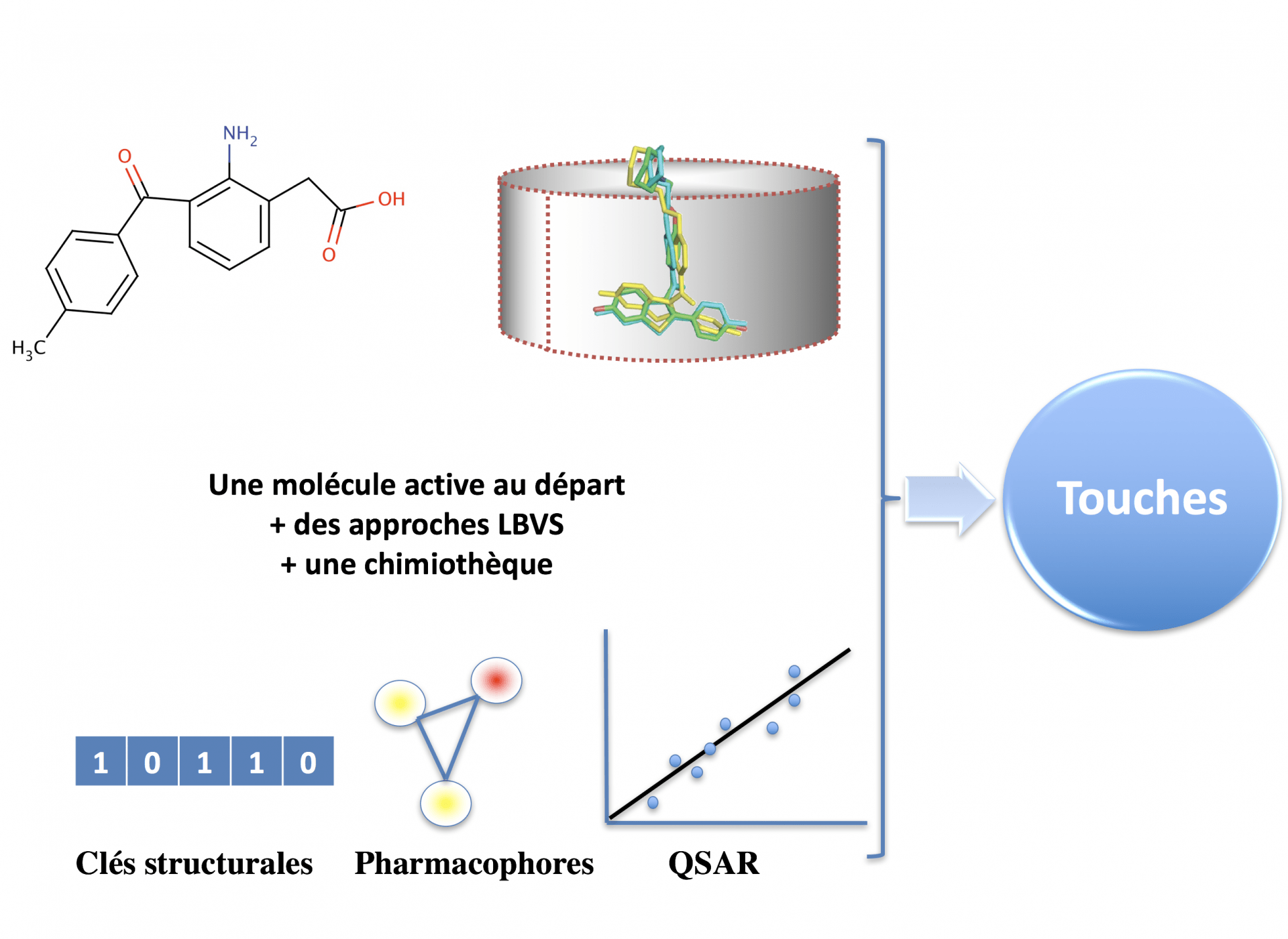
Elles reposent sur l’utilisation de descripteurs (topologie, propriétés quantiques, taille, lipophilie, etc.) ou de sous-structures chimiques, véritable carte d’identité d’une petite molécule. D’autres approches qui comparent la forme 3D des molécules existent. Nous n’aborderons pas ces méthodes dans cet article.
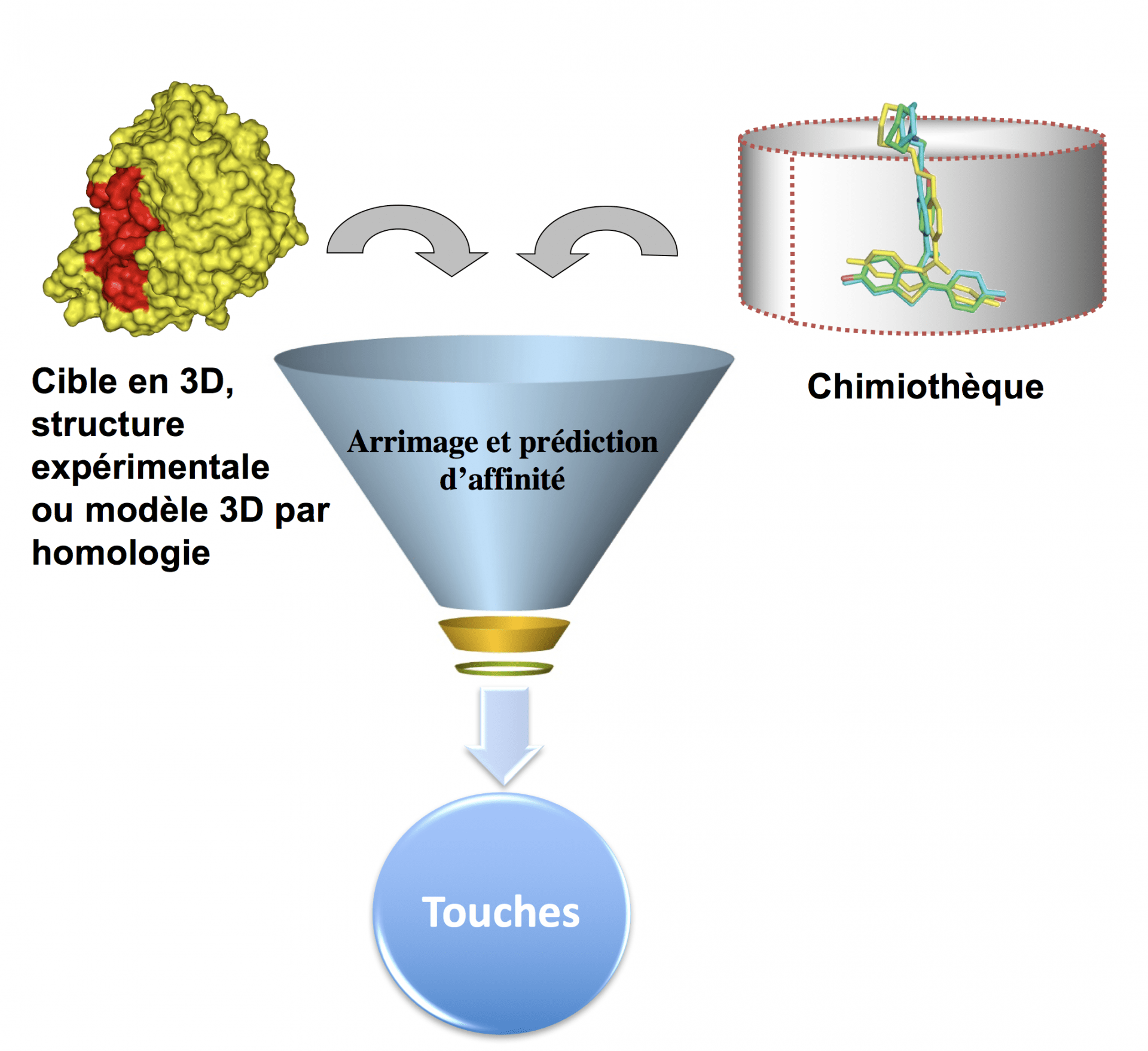
Chaque composé chimique présent dans la chimiothèque est placé (« docking » ou ancrage) dans le site actif de la cible (en rouge). Le docking à haut débit consiste ainsi à prédire à la fois la conformation active et l'orientation relative de chacune des molécules de la chimiothèque sélectionnée par rapport à la cible d'intérêt. Une fonction mathématique va évaluer la complémentarité géométrique et chimique des partenaires afin d’estimer l’affinité (« scoring »).
Bien que les calculs comportent encore des imprécisions, les résultats du criblage virtuel sont en général pertinents et supérieurs dans de nombreux cas à ceux obtenus par les techniques de criblage expérimental. Dans l’absolu, les deux types de criblage, expérimental et virtuel, sont complémentaires ; néanmoins, les restrictions budgétaires et la puissance des approches chémoinformatiques en cours de développement devraient imposer dans un avenir proche l’utilisation systématique de ces outils in silico en amont des tests expérimentaux. Les nouveaux algorithmes d’intelligence artificielle et la puissance des nouveaux ordinateurs poussent aussi dans cette direction. Il est important de souligner à nouveau qu’identifier des touches actives est une étape nécessaire mais non suffisante. Comme nous l’avons vu précédemment, une touche doit être optimisée à plusieurs niveaux : affinité et sélectivité pour la cible thérapeutique, diminution de la toxicité et amélioration des paramètres pharmacocinétiques. Là encore, deux options sont possibles, hasard et intuition ou bien utilisation d’approches plus rationnelles de type QSAR (« Quantitative Structure Activity Relationship », voir ci-après), intelligence artificielle, optimisations multiparamétriques ou de type SBVS-LBVS, couplées à des études biophysiques et à de la chimie médicinale.
Ligand-based virtual screening (LBVS)
Il existe plusieurs approches de LBVS, dont l’objectif est de rechercher, dans une base de données chimiques, des molécules similaires à une molécule bioactive déjà identifiée. Une première approche consiste à utiliser la notion d’empreintes moléculaires (ou « fingerprint »). Un « fingerprint » est un ensemble de descripteurs, généralement représenté sous la forme d’un vecteur de bits (les éléments du vecteur prenant la valeur 0 ou 1) : voir figure 10. Chaque élément du vecteur représente une propriété, typiquement la présence ou non d’un motif chimique. Un score est ensuite calculé pour comparer les composés. La similarité entre deux molécules (la comparaison de l’empreinte moléculaire de 2 molécules) est généralement évaluée en calculant le coefficient de Tanimoto (Tc) suivant la formule : Tc = nE/(nA + nB - nE). nE est le nombre de bits communs (nombre de sous-structures chimiques communes) entre les deux structures, nA est le nombre de sous-structures identifiées dans le composé A, et nB, le nombre de sous-structures identifiées dans le composé B. Quand les molécules sont proches, le score de chaque bit est proche de 1 ou égal à 1, ; quand elles sont très différentes, le score est proche de zéro.
L’autre grande approche LBVS est le criblage à l’aide de modèles pharmacophores. Le concept de pharmacophore a été développé par Ehrlich à la fin du XIXème siècle. La définition officielle de l’IUPAC (International Union of Pure and Applied Chemistry) datant de 1998 indique que l’ensemble des propriétés stériques et électroniques d’une molécule, nécessaires pour assurer l’établissement d’interactions supramoléculaires optimales avec une cible biologique spécifique et engendrer ou bloquer une réponse biologique, constitue un pharmacophore. D’après cette définition, des molécules partageant le même pharmacophore pour une cible donnée devraient donc se lier de manière identique à ce récepteur et présenter des profils d’activité similaires. Le pharmacophore généré à partir de molécules actives sur notre cible est donc utilisé in silico pour cribler une chimiothèque à la recherche de molécules se superposant à ce pharmacophore.
Les approches QSAR (« Quantitative Structure Activity Relationship ») représentent une autre manière de rechercher des molécules dans une chimiothèque. Ces techniques permettent de relier par une relation mathématique les descripteurs chimiques, soit à l’activité biologique dans une approche de relation structure-activité quantitative, soit à une propriété (physico-chimique ou pharmacocinétique) dans le cas des relations structure-propriété quantitative (QSPR pour quantitative structure property relationship). De tels modèles statistiques, basés sur des descripteurs moléculaires calculés, sont le point de départ de nombreux processus de sélection de molécules. Ces modèles sont souvent construits à partir d’un jeu de référence, appelé jeu d’apprentissage, permettant de sélectionner le(s) descripteur(s) le(s) plus adapté(s) dans la construction d’un modèle. Les modes de construction de ces modèles peuvent être simples (régression linéaire…) ou plus sophistiqués (algorithmes génétiques, réseaux de neurones, forêts aléatoires…). De nos jours, d’autres algorithmes peuvent etre utilisés comme les réseaux de neurones convolutifs (exemple de « deeplearning » souvent confondu avec l’intelligence artificielle).
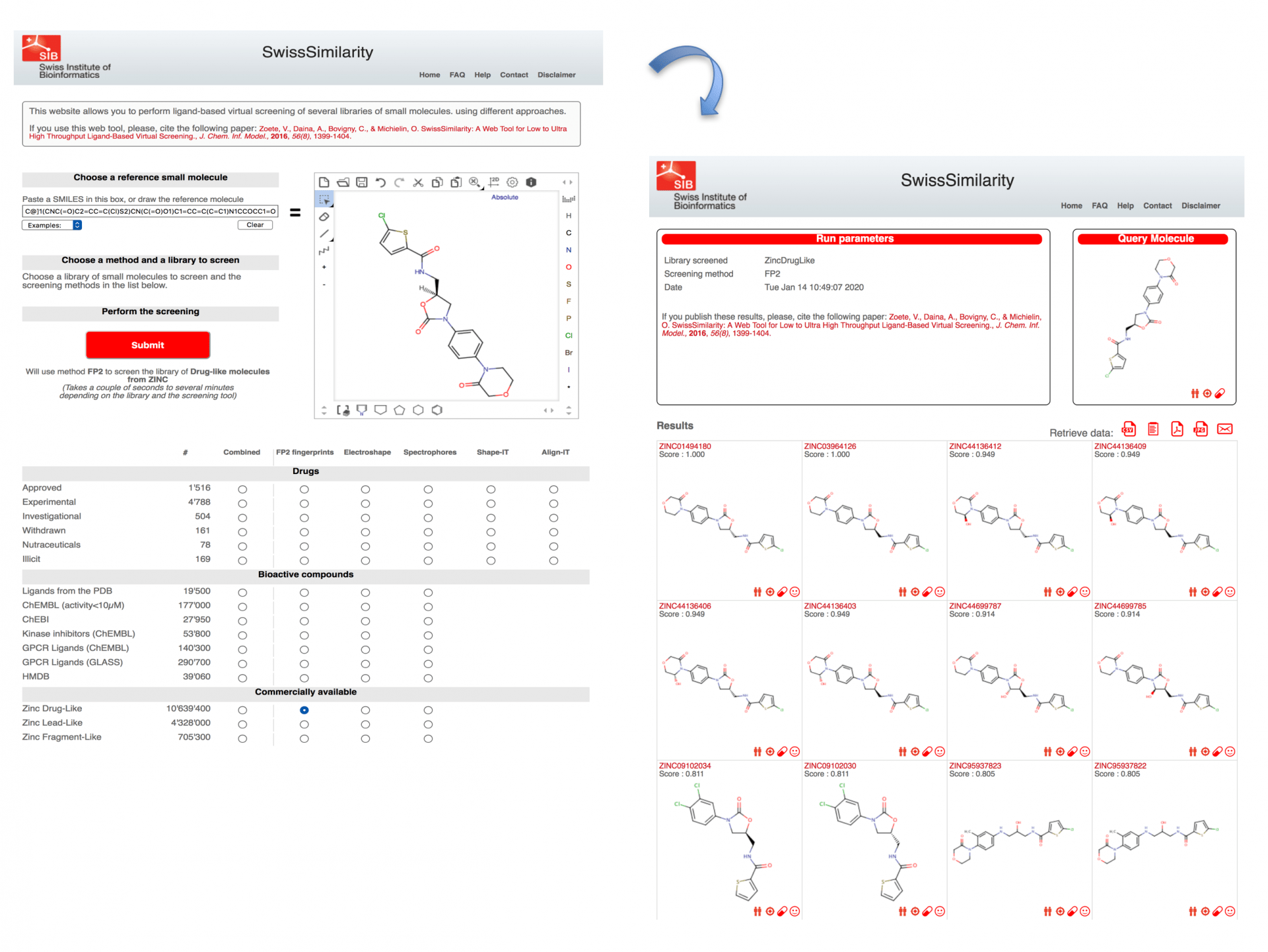
De nombreux services en ligne permettent de faire du criblage virtuel. Par exemple, pour ce qui concerne le criblage LBVS, il est possible d’utiliser SwissSimilarity. Dans ce cas, il faut comme point de départ une molécule qui est active sur la cible, ou que l’on suppose active. Revenons sur l’inhibiteur du facteur X, le rivaroxaban. Il est possible d’obtenir la structure chimique de ce composé sur DrugBank par exemple : dans la section Structure, « download », choisir le format SMILES :
[H][C@]1(CNC(=O)C2=CC=C(Cl)S2)CN(C(=O)O1)C1=CC=C(C=C1)N1CCOCC1=O
Il faut ensuite copier et coller cette formule chimique dans SwissSimilarity, et sélectionner une chimiothèque déjà préparée sur le site pour rechercher des molécules similaires. Ces molécules proches de la molécule de départ, ici le rivaroxaban, ont en théorie de fortes chances d’être aussi actives sur le facteur X, sachant qu’en général, quand des molécules sont très similaires elles ont souvent la même activité biologique (c’est le principe de similarité). Il faut également choisir l’algorithme de recherche : la méthode FP2 (empreinte moléculaire) est ici sélectionnée et la base de données peut être la « Zinc Drug-like » qui contient plus de 10 millions de molécules généralement disponibles chez les vendeurs de produits chimiques (Fig. 5).
Structure-based virtual screening (SBVS)
Lorsque la structure 3D de la cible biologique d’intérêt est disponible, les méthodes basées sur la structure du récepteur ou cible peuvent être employées pour réaliser le criblage virtuel. Dans un premier temps, le logiciel positionne les petites molécules chimiques déjà générées en 3D dans une poche ou sur toute la surface de la cible. Un score sera ensuite calculé pour essayer de prédire l’affinité des composés envers la cible. Il sera alors possible de classer les molécules par ce score théorique. Dans la pratique, plusieurs points sont à souligner. Les cibles et les petites molécules sont généralement flexibles. Pour aller plus vite, quand il y a des millions de molécules à traiter, on suppose que la cible est rigide et, dans ce cas, seule la flexibilité des ligands est explorée. Il est possible de préparer plusieurs conformations de la cible (par exemple par dynamique moléculaire) et de faire du « docking » sur les différentes structures 3D de la cible et de fusionner ensuite les résultats. Ensuite, il faut savoir que l’étape de « scoring » est une étape sensible, les scores calculés ne sont en général pas très précis, et il faut donc utiliser plusieurs approches dites de « post-processing » qui impliquent des calculs d’énergie d’interactions cible-ligands beaucoup plus longs et complexes. Les forces ou paramètres sous-jacents à la liaison d’un ligand à son récepteur sont d’une part la complémentarité de forme, et d’autre part l’établissement d’interactions entre le ligand et le site de liaison, que ce soit des liaisons hydrogènes, des interactions électrostatiques et des interactions de Van der Waals ou encore des interactions hydrophobes. Classiquement, on trouve trois grands types de fonctions de score : les fonctions de scores basées sur les champs de force, les fonctions de scores empiriques (qui tentent de reproduire des données expérimentales d’énergies de liaison), et les fonctions de scores basées sur les connaissances (statistiques). De nouvelles fonctions de score sont à présent disponibles, elles ont été développées avec des méthodes d’apprentissage et entraînées sur des nouveaux jeux de données. Le choix d’une fonction de score est critique, et actuellement aucune fonction de score existante n’est parfaite car il y a un certain nombre d’approximations. De plus, les molécules d’eau ne sont généralement pas traitées. Malgré ces problèmes, il est pourtant possible de sélectionner les molécules avec ces fonctions mathématiques. Néanmoins, les experts du domaine ne font jamais pleinement confiance à une fonction de score, ils en utilisent plusieurs et analysent sur l’écran de l’ordinateur à l’aide de logiciels de graphique moléculaire les complexes cible-ligands prédits.
Plusieurs services en ligne permettent de faire du criblage de type SBVS, notamment MTiOpenScreen. Sur ce serveur, l’utilisateur doit fournir le fichier PDB de sa cible. On peut trouver sur la PDB, la structure d’un inhibiteur co-cristallisé avec le facteur X, dont le code est « 2w26 ». Il est possible d’éditer ce fichier et d’enlever la petite molécule afin de générer un fichier qui ne contient que l’information sur la protéine. On le télécharge sur MTiOpenScreen (Fig. 6) et on peut ensuite sélectionner une chimiothèque, dans le cas présent, Drugs-lib, qui contient environ 4000 médicaments connus.
La zone de recherche pour le « docking » est définie par le paramètre « Grid calculation » et on choisit le mode « list of residues ». Plus bas, on ajoute dans la petite boîte sur l’interface le nom de deux acides aminés du site catalytique, l’histidine 57 et la sérine 195.
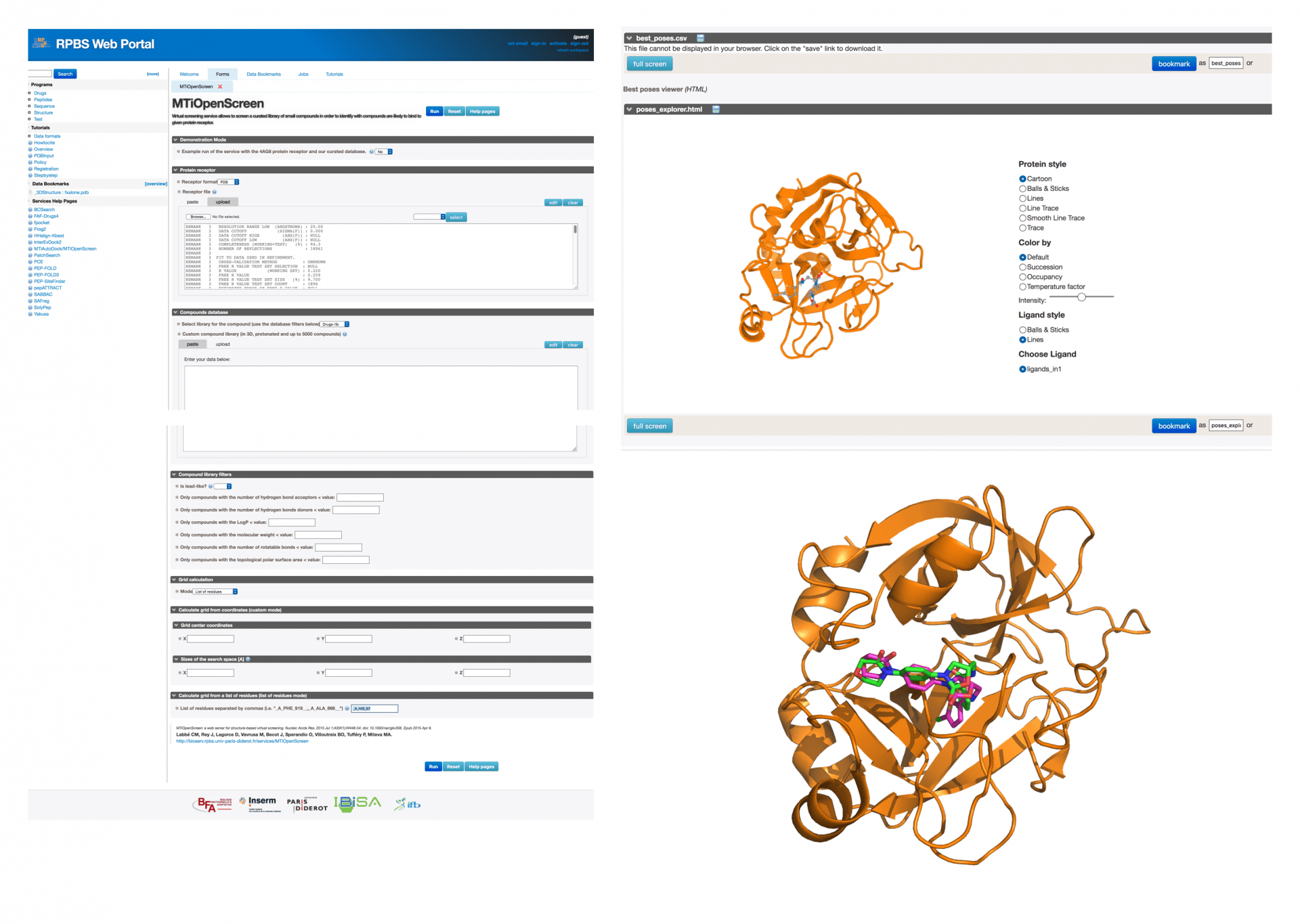
L’utilisateur donne la structure 3D de sa cible et peut arrimer des molécules déjà préparées ou ses propres molécules. Il est possible de visualiser les molécules positionnées dans la poche par le calcul théorique. Dans ce cas, le « docking » est fait avec le logiciel AutoDock Vina. On peut voir sur la figure à droite, la protéine, le facteur X (en orange), avec son inhibiteur arrimé par le calcul (en vert) avec la structure expérimentale (en violet).
Ces résidus sont connus, on peut les identifier sur la PDB, sur UniProt ou dans PubMed. Dans la case : « Calculate grid from a list of residues (list of residues mode) », nous ajoutons en suivant scrupuleusement les indications, un code un peu spécial qui définit ces deux acides aminés : _A_HIS_57__,_A_SER_195__.
Dans cet exemple, on cherche des molécules qui sont déjà des médicaments et qui agissent par exemple sur d’autres systèmes biologiques que la coagulation afin de voir s’il est possible de repositionner une molécule sur une autre cible et lui trouver une autre application thérapeutique. De plus, on doit pouvoir aussi évaluer la performance des méthodes : on doit retrouver ici le rivaroxaban dans les molécules qui ont une bonne affinité prédite pour le facteur X.
Il est possible de visualiser les meilleures molécules positionnées dans le facteur X en ligne, mais, l’utilisateur avancé préférera télécharger le fichier de résultats et analyser les données sur sa machine, avec un logiciel de graphisme moléculaire plus sophistiqué comme Chimera. Les approches par « docking » nécessitent des calculs plus complexes que ceux vu ci-dessus dans l’exemple du LBVS. Ainsi, plusieurs minutes ou heures de calcul sont souvent nécessaires pour obtenir les résultats, en fonction de la charge du cluster.
Conclusion
Aujourd’hui Internet propose une manne de données numériques utiles à la recherche de nouveaux médicaments. La source semble quasiment intarissable : en moyenne, toutes les 2 semaines dans le monde, une base de données ou un logiciel est publié dans le domaine. Ces outils sont pour la plupart performants mais, comme ils sont implémentés en ligne, toutes les options ne sont pas toujours disponibles dans l’interface. Ces outils en ligne facilitent néanmoins la conception de molécules thérapeutiques en aidant le chercheur à tester ses hypothèses plus rapidement, lui permettant ainsi de concentrer ses efforts sur les options les plus probables. Ces services sont relativement simples à utiliser (c’est bien sûr un de leurs objectifs), mais il faut souligner que les concepts et algorithmes manipulés en arrière-plan sont souvent très complexes (des mois voire des années d’optimisation et de développement sont souvent nécessaires pour mettre en place une base de données ou coder un nouvel outil). Par conséquent, nul ne peut se dispenser d’un temps d’apprentissage, de lecture et d’entraînement avec les tutos/démos/vidéos qui accompagnent ces outils et ressources. Nous avons ici seulement survolé un petit nombre de bases et d’outils parmi les milliers existants, sans compter ceux qui concernent les approches pour les produits biologiques et les biomédicaments.
Il faut considérer ces bases de données et outils en ligne comme une vraie vitrine de compétences pour un pays. Les domaines de recherche associés à la recherche de médicaments sont dits « aux interfaces » car ils font appel à différentes disciplines scientifiques telles que la biologie, la chimie, la médecine, l’informatique ou encore la biophysique. Souvent malmenées en raison de leur positionnement stratégique aux interfaces entre différents départements, ministères, groupes décisionnels... ces approches in silico sont pourtant importantes non seulement pour la recherche scientifique mais aussi pour l’enseignement. Il serait donc important de définir une politique de recherche qui prenne en compte leurs spécificités. Plusieurs universités ou lycées dans le monde utilisent des bases de données et logiciels gratuits pour illustrer le développement des médicaments ou pour enseigner la chimie et la biologie (Rodrigues et col. J. Chem. Educ. 2015, 92, 5, 827-835; Daina et col., Chimia (Aarau). 2018, 72:55-61; Villoutreix et col., Drug Discovery Today, 2013, 18 :1081-9; Sydow et col., J Cheminform. 2019, 11: 29). En associant ces différents outils, l’enseignement ou l’apprentissage d’une matière devient moins aride et plus pragmatique.
Les défis à relever… dans 10, 20, 50 ans ? Dans les années qui viennent de nombreux progrès sont à espérer en matière de prédiction de toxicité, de simulation virtuelle du voyage des molécules dans le corps humain, de simulation d’interactions entre les molécules, via des arrimages in silico et des approches d’intelligence artificielle, de navigation dans l’espace des cibles thérapeutiques et dans l’espace chimique... La recherche in silico des effets des composés chimiques ou de certains biomédicaments ou du couplage petite molécule-biomolécule (par exemple, les immunoconjugués) non plus sur une cible thérapeutique particulière mais sur l’ensemble d’un système biologique est déjà en marche. Les résultats sont encourageants, autorisant pour la première fois la compréhension fine de certains mécanismes moléculaires responsables d’effets secondaires hors de portée des approches expérimentales dans certains cas. Les travaux sur les réseaux d’interaction et les avancées théoriques devraient aboutir à la création d’organes virtuels et à la conception de molécules innovantes entièrement conçues sur ordinateur et par des robots avec une réduction concomitante de l’expérimentation animale tout en étant mieux adaptées au profil des patients.
Bibliographie
En plus des références mentionnées dans le texte, le lecteur est invité à consulter les ressources ci-dessous, rédigées en français.
- Vayer P, Arrault A, Lesur B, Bertrand M, Walther B. Apports de la chémoinformatique dans la recherche et l’optimisation des molécules d’intérêt thérapeutique. Med Sci (Paris), 2009, 25:871-7
- Bureau R. Modélisation moléculaire et conception de nouveaux ligands d'intérêts biologiques. Techniques de l’ingénieur 2014, 1-19.
- Rognan D, Bonnet P. Les chimiothèques et le criblage virtuel. Med Sci (Paris). 2014, 30:1152-60
- Sperandio O, Villoutreix B., Morelli X, Roche P. Les chimiothèques ciblant les interactions protéine-protéine. Med Sci (Paris). 2015, 31:312-9.
- Maupetit J, Saladin A, Tuffery P. Prédiction en ligne de la structure des protéines. SPECTRA ANALYSE 2010, 276: 27-33
Remerciements
L'auteur remercie Natacha Oliveira pour sa relecture attentive et ses tutos/vidéos « DataWarrior ».
Documents à télécharger