Les effets isotopiques sont à l'origine d'applications dans des domaines très variés de la chimie. Ils permettent par exemple d'interpréter l'enrichissement isotopique observé dans les processus biologiques, phénomène mis à profit pour effectuer les analyses chimiques d'authentification de produits naturels et synthétiques. Les effets isotopiques permettent également d'étudier dans le détail les mécanismes réactionnels.
Introduction
Les effets isotopiques sont à l'origine d'applications dans des domaines très variés de la chimie. Ils permettent par exemple d'interpréter l'enrichissement isotopique observé dans les processus biologiques, phénomène mis à profit pour effectuer les analyses chimiques d'authentification de produits naturels et synthétiques (voir les articles l'isotope : traceur d'origine. Distribution isotopique dans les composés naturels et Contribution des analyses isotopiques à la lutte contre les trafics de drogue). Les effets isotopiques permettent également d'étudier dans le détail les mécanismes réactionnels. Cet article propose une introduction aux effets isotopiques cinétiques : après avoir donné quelques définitions de bases, nous présenterons les effets isotopiques cinétiques les plus utilisés ainsi que leur application à la détermination de mécanisme.
Définitions
Isotopes
Les éléments chimiques qui constituent la matière sont tous bâtis sur le même modèle : l'atome est constitué d'un noyau composé de protons, chargés positivement, et de neutrons, électriquement neutres. Autour de ce noyau gravitent des électrons, particules chargées négativement. Le nombre de protons est égal au nombre d'électrons, ce qui assure la neutralité de l'édifice atomique. Ce nombre est appelé le numéro atomique, noté Z, et il est caractéristique de l'élément chimique. Toutefois, pour un même élément chimique, il peut exister différents noyaux : en effet, si le nombre de protons est toujours égal à Z, le nombre de neutrons peut varier. On parle alors d'isotopes de l'élément chimique. Les isotopes sont caractérisés par le nombre de masse, noté A, égal à la somme des nombres de protons et de neutrons ; on différencie les isotopes d'un élément X par la notation AX. Par exemple, le noyau de l'atome d'hydrogène est constitué d'un proton qui peut, à l'état naturel, être accompagné de zéro, un ou deux neutron(s). L'hydrogène existe donc sous trois formes isotopiques 1H (noté simplement H), 2H (appelé deutérium, noté D) et 3H (appelé tritium, noté T).
Effet isotopique cinétique
Si les différents isotopes d'un même élément peuvent, en première approximation, être considérés comme présentant les mêmes propriétés du point de vue de la réaction chimique, un examen plus poussé révèle que la substitution d'un atome par un de ses isotopes produit des effets perceptibles thermodynamiquement et cinétiquement. Le principe de l'effet isotopique cinétique est la comparaison d'une même réaction chimique avec deux substrats se différenciant par une modification isotopique. La perturbation induite sur le système, quoique faible, est perceptible cinétiquement, elle se traduit par une différence entre les constantes cinétiques de réaction kI associées aux différents isotopes I. Le phénomène est observable avec les éléments les plus courants : H, C, N, O, P, S,... Dans la suite, on considérera des substitutions isotopiques de l'hydrogène H par le deutérium D : ce sont celles qui présentent les effets cinétiques les plus intenses et qui sont donc les plus utilisées pour étudier les mécanismes réactionnels.
La substitution isotopique ne perturbe pas les termes énergétiques de nature électronique, mais seulement les termes énergétiques liés à la masse des atomes (c'est-à-dire les niveaux vibrationnels et rotationnels mais pas les niveaux électroniques). Chaque fréquence de vibration d'une liaison est liée à la valeur de la masse atomique liés (de manière différente selon la nature des vibrations : élongation, déformation, ...) et varie avec l'isotope considéré. Au cours d'une réaction chimique l'énergie vibrationnelle du système en réaction varie lors du passage de l'état initial à l'état de transition, puisqu'une ou plusieurs liaisons sont cassées lors de la réaction chimique (lorsqu'une liaison s'allonge, la force de la liaison diminue donc la fréquence de la vibration associée varie ; elle diminue dans le cas d'une vibration d'élongation). Le remplacement d'un atome par un de ses isotopes produit un changement des niveaux d'énergie vibrationnelle dans l'état fondamental et éventuellement, dans l'état de transition.
- Si la liaison molécule-isotope n'est pas affectée par la réaction ou si le changement ne se produit pas au cours de l'étape cinétiquement déterminante, il n'y a pas d'effet isotopique : kH/kD = 1.
- Si la liaison molécule-isotope est affectée durant l'étape cinétiquement déterminante, il y a effet isotopique : kH/kD ≠ 1 ; kH/kD est d'intensité variable et peut être supérieur ou inférieur à 1.
Effet isotopique primaire (EIP)
Définition
On observe un EIP lorsque la liaison impliquant l'atome d'hydrogène substitué par le deutérium est rompue au cours de l'étape cinétiquement déterminante.
Étude théorique
L'étude théorique de l'EIP modélise la liaison selon l'approximation de l'oscillateur harmonique : on considère la vibration le long de l'axe Ox de deux masses mA et mB reliées par un ressort de constante de raideur K. Alors la fréquence ν de la vibration est donnée par la loi de Hooke où μ est la masse réduite du système, μ = m1 m2 / (m1 + m2) :
ν = 1/2π √(K/μ)
La modification de la fréquence de vibration par substitution isotopique est calculable en posant comme hypothèse que les constantes de force des liaisons C-H et C-D sont identiques, ce qui est justifié puisque les propriétés de la liaison sont dues aux interactions entre particules chargées du système (électrons et noyaux), et sont donc insensibles à une variation du nombre de neutrons. Alors μD/μH ≈ 2 et vH/νD ≈ √2
Ce phénomène est visible en spectroscopie infrarouge en examinant les vibrations d'élongation des liaisons C-H et C-D. On observe des nombres d'onde1 ν(CHCl3) ≈ 3000 cm-1 et ν(CDCl3) ≈ 2100 cm-1 dont le rapport est voisin de √2.
La mécanique quantique montre que l'énergie vibrationnelle des liaisons est quantifiée. Elle ne peut prendre que des valeurs discrètes En, n entier, qui sont reliées à la fréquence de vibration ν par la relation suivante où h est la constante de Planck :
En = (n + ½) hν
Le niveau d'énergie le plus bas, E0, est associé à la valeur n = 0. Il est appelé EPZ (Énergie de Point Zéro) et sa valeur est :
E0 = ½ hν.
Comme ν = 1/2π √(K/μ) et K est considérée comme constante en passant de C-H à C-D, les oscillateurs harmoniques C-H et C-D ont des EPZ différentes ; comme μD > μH, il vient E0(A-H) > E0(A-D). Cette différence d'énergie des EPZ est à l'origine de l'effet isotopique primaire. En effet, la différence entre les énergies de dissociation de C-D et C-H est ED(A-D) - ED(A-H) = E0(A-H) - E0(A-D) > 0, donc la dissociation de C-H est plus facile que la dissociation de C-D, toutes conditions égales par ailleurs. De cette différence énergétique, il est possible de passer à une différence cinétique. Un modèle complet utilisant la thermodynamique statistique permet d'obtenir une valeur théorique kH/kD = 8,8 à 298 K et on montre que le rapport kH/kD est une fonction décroissante de la température [1]. Dans la pratique, on mesure des valeurs comprises entre 1 et cette valeur théorique de 8,8. L'écart à la valeur théorique donne des informations sur la structure de l'état de transition2.
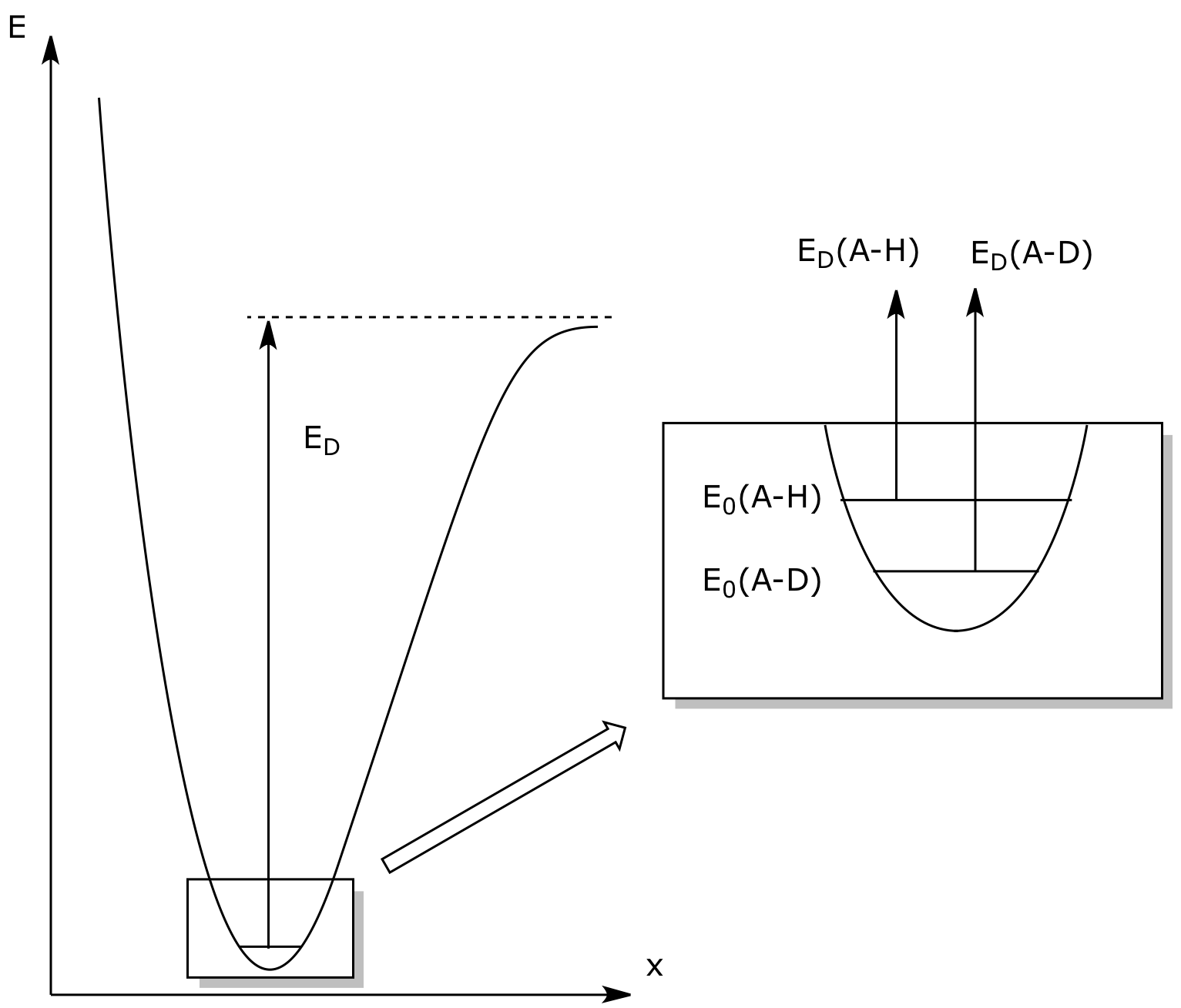
ED : énergie de dissociation de la liaison A-H ou A-D. La différence ED(A-D) - ED(A-H) est égale à la différence entre les EPZ : ED(A-D) - ED(A-H) = E0(A-H) - E0(A-D).
Application à la détermination de mécanisme
Application à la détermination de l'étape cinétiquement déterminante : exemple de la bromation des cétones en milieu acide
L'EIP a permis de déterminer les caractéristiques du mécanisme suivant postulé pour la bromation des cétones en milieu acide donné à la figure 2 :
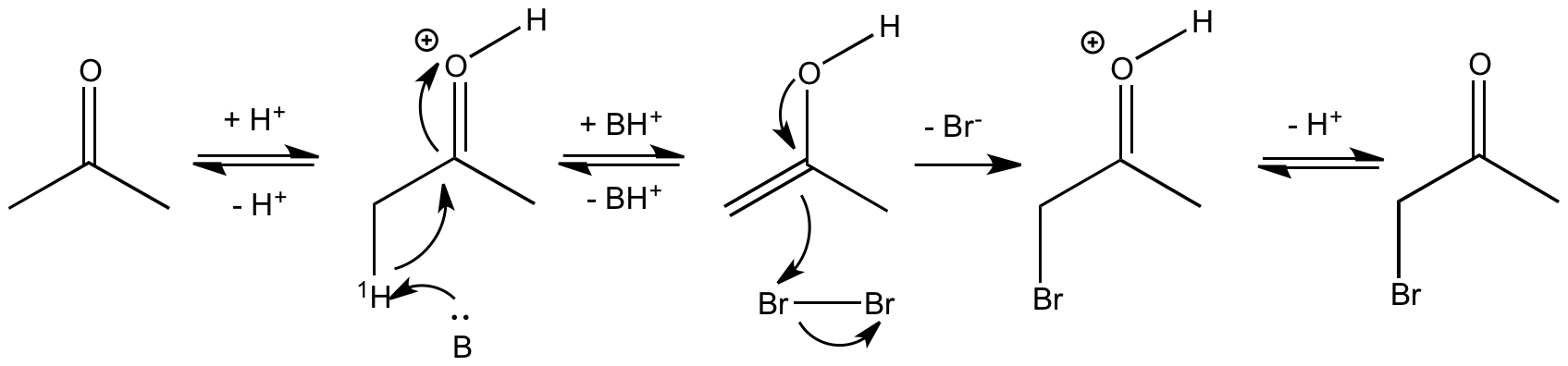
La première étape est un équilibre acido-basique de protonation de l'oxygène du carbonyle. Elle est suivie de l'énolisation par arrachage du proton en α du carbonyle (noté 1H). On observe ensuite l'addition électrophile du brome sur la double liaison énolique puis un nouvel équilibre acido-basique qui déprotone l'oxygène du carbonyle. Cette dernière étape permet de régénérer le proton qui sert ici de catalyseur.
La substitution isotopique de l'hydrogène en β du carbone du carbonyle (noté 1H) par le deutérium conduit à une diminution très nette de la vitesse d'énolisation : kH/kD = 7. Cet EIP permet de conclure que la rupture de la liaison C-H a lieu pendant l'étape cinétiquement déterminante (voir le mode opératoire [5]).
Application à la validation d'un mécanisme réactionnel : exemple de la substitution électrophile aromatique
Deux mécanismes, donnés à la figure 3, ont été proposés pour la nitration du benzène (substitution électrophile aromatique de l'hydrogène par le groupement nitro). La voie A est un mécanisme en une étape : la liaison C-N est formé simultanément à la rupture de la liaison C-H. La voie B se déroule en deux étapes. Dans un premier temps, on observe la formation de la liaison C-N tandis que la liaison C-H reste intacte. Cette étape mène à un intermédiaire réactionnel au sein duquel la charge positive est délocalisée sur le cycle benzénique (intermédiaire de Wheland aussi appelé complexe σ). On observe ensuite la rupture de la liaison C-H lors d'une étape dite de « réaromatisation ».
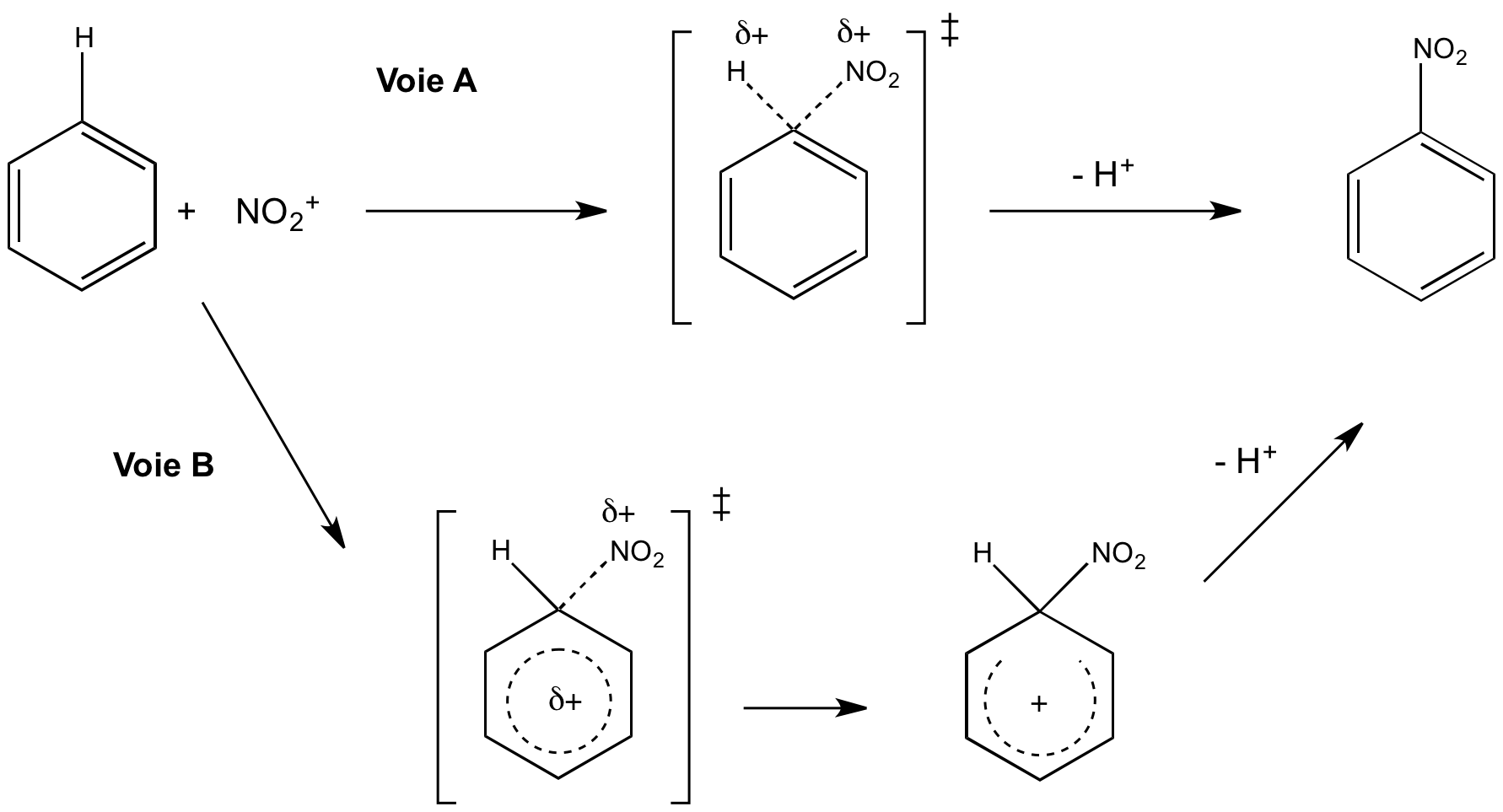
La voie A se déroule en une étape : il y a rupture et formation concertée des liaisons C-H et C-N respectivement. La voie B se déroule en deux étapes : formation de la liaison C-N puis rupture de la liaison C-H.
Historiquement, la voie A était privilégiée par des arguments énergétiques : elle permet de conserver l'aromaticité du noyau benzénique1 pendant tout le déroulement de la réaction, contrairement à la voie B qui voit la rupture d'une des liaisons σ. Pourtant cette hypothèse a été invalidée par l'absence d'EIP. En effet, expérimentalement on mesure kH/kD = 1,0. Cela permet d'éliminer avec certitude la voie A : elle n'est constitué que d'une étape, donc le début de rupture de liaison C-H postulé devrait se traduire par un EIP mesurable (kH/kD > 1). Cela prouve que cette rupture a lieu au cours d'une autre étape et qu'elle n'est pas cinétiquement déterminante. L'EIP permet ici de conclure quant à la nature réelle du mécanisme.
Il a par la suite été montré que les autres substitutions électrophiles aromatiques (halogénation, acylation, sulfonation, couplage diazoïque...) se déroulent suivant le même mécanisme que la nitration. Toutefois la nature de l'étape cinétiquement déterminante est propre à chaque réaction. Pour les réactions de nitration et d'halogénation du benzène, l'attaque électrophile est cinétiquement déterminante tandis que c'est la réaromatisation pour l'acylation, la sulfonation et le couplage diazoïque [3].
Effet isotopique secondaire (EIS)
Définition
L'effet isotopique est dit secondaire lorsque la liaison examinée, celle où la substitution isotopique est effectuée, n'est pas cassée mais seulement perturbée (allongement, raccourcissement...) au cours de l'étape cinétiquement déterminante de la réaction. L'intensité de cet effet est plus faible que celle de l'EIP, c'est pourquoi il n'est en pratique observé que pour les liaisons C-H. Selon la position de la liaison examinée par rapport au centre réactionnel, on parle d'EIS α (H est lié au carbone site de la réaction) ou β (H est séparé du site de la réaction par deux liaisons).
Effet isotopique secondaire α
L'EIS α permet de mettre en évidence un changement de géométrie du carbone au cours du passage état initial → état de transition. Il se subdivise en deux cas : l'effet normal et l'effet inverse.
- L'EIS α est dit normal si kH/kD > 1. On observe alors le changement carbone tétragonal → trigonal plan.
- L'EIS α est dit inverse si kH/kD < 1. On observe alors le changement d'hybridation carbone trigonal plan → tétragonal.
Application à l'établissement d'un mécanisme réactionnel : exemple de la formation des cyanhydrines
La formation des cyanhydrines est une réaction qui présente un EIS α inverse. Par exemple kH/kD= 0,73 pour la réaction de la figure 4. Cette donnée permet de proposer le mécanisme de la figure 4 où l'étape cinétiquement déterminante est l'attaque de l'ion cyanure sur le carbonyle. C'est le passage d'un carbone trigonal plan à un carbone tétragonal dans cette étape qui est à l'origine de l'EIS α inverse mesuré par substitution de l'hydrogène lié à ce carbone (noté 1H).
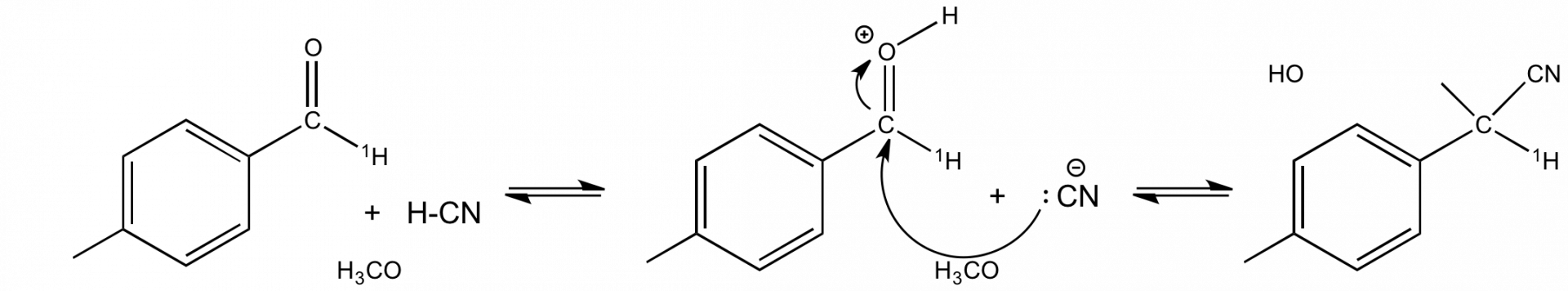
Application à l'établissement d'un mécanisme réactionnel : exemple de l'hydrolyse du chlorure de benzyle
La réaction d'hydrolyse du chlorure benzylique présente un EIS α normal avec kH/kD = 1,3. Cela permet de proposer le mécanisme de la figure 5 où l'étape cinétiquement déterminante est la formation du carbocation. C'est le passage d'un carbone tétragonal à un carbone trigonal plan dans cette étape qui est à l'origine de l'EIS α normal mesuré par substitution de l'hydrogène lié à ce carbone (noté 1H).
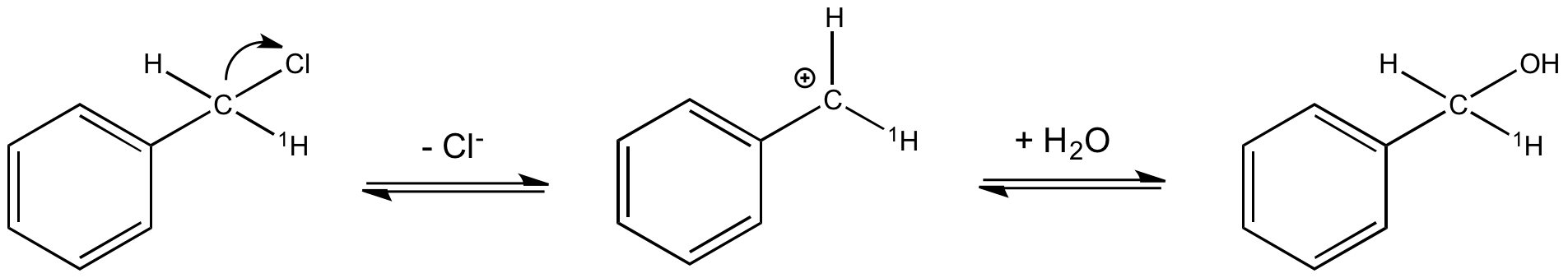
Effet isotopique secondaire β
L'effet isotopique secondaire β renseigne sur la stabilisation à travers l'espace d'un cation : l'hyperconjugaison. Cette stabilisation cause un affaiblissement de la liaison C-H. C-H est plus affecté que C-D car la liaison C-H est plus polarisable que C-D. Cela se traduit par un rapport kH/kD > 1, mais toujours faible en intensité : 1,2 au maximum.
L'EIS β dépend de plusieurs facteurs comme l'électronégativité des substituants et la géométrie du système. Il a été exploité en particulier pour étudier les mécanismes de substitutions nucléophiles [3].
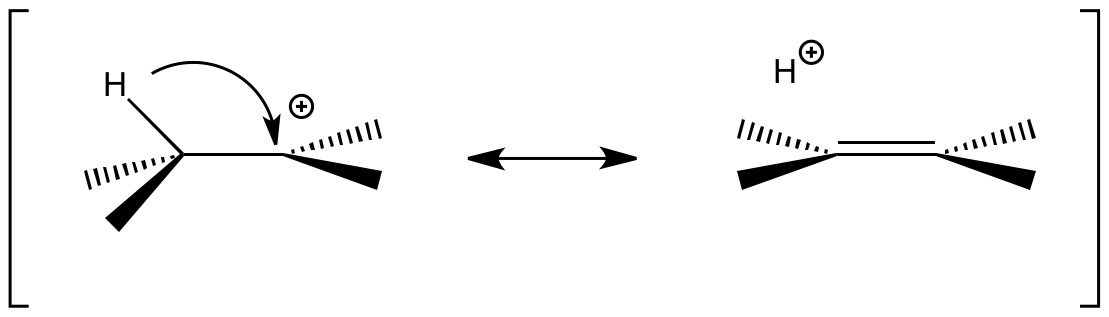
L'écriture des formes mésomères permet d'illustrer l'affaiblissement de la liaison C-H provoqué par la délocalisation électronique de ses électrons vers le carbocation.
Conclusion
Nous avons vu sur quelques exemples tirés de la chimie organique l'exploitation qui pouvait être faite des effets isotopiques primaire et secondaire pour déterminer les mécanismes réactionnels. Ces effets sont également mis à profit en biochimie pour élucider les mécanismes catalysés par les enzymes par exemple [4]. Toutefois, dans la pratique, l'interprétation des mesures cinétiques peut être très difficile, par exemple un rapport kH/kD compris entre 1 et 2 peut être attribué à un EIP ou un EIS. C'est pourquoi cette méthode est en général couplée à d'autres mesures : spectroscopiques, électrochimiques...
Bibliographie
De nombreux exemples de réactions présentant un effet isotopique cinétique sont donnés dans les références 1 à 3. Les références 2 et 3 proposent en plus une approche théorique complète de l'EIP et de l'EIS. La référence 4 évoque l'utilisation des effets isotopiques cinétiques en biochimie. La référence 5 propose un lien vers une manipulation permettant de mesurer l'EIP dans la réaction de bromation de la propanone.