Ce premier article du dossier Conception et repositionnement de médicaments rappelle les différentes phases de la « vie » d'un médicament (dont le principe actif est d'origine chimique)1, de la découverte du principe actif à la commercialisation, ainsi que les enjeux et contraintes liées à la recherche pharmaceutique.
Qu'est-ce-qu'un médicament ?
Le code de la Santé publique (article L.5111-1) définit ainsi le médicament : « toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l’égard des maladies humaines ou animales, ainsi que toute substance ou composition pouvant être utilisée chez l’homme ou chez l’animal ou pouvant leur être administrée, en vue d’établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions physiologiques en exerçant une action pharmacologique, immunologique ou métabolique. »
Un médicament contient :
- un principe actif, substance d’origine chimique ou naturelle qui agit contre une maladie, soit en la traitant, soit en limitant son aggravation ;
- des excipients, substances d’origine chimique ou naturelle qui facilitent l’utilisation du médicament mais ne présentent pas d’effet curatif ou préventif. Parmi les excipients on trouve des arômes, des sucres, des substances permettant d'obtenir une forme facilement administrable au patient (comprimé, gélule, sirop, solution injectable...), des conservateurs.
Chronologie de la vie d'un médicament
Élaborer un médicament est un processus long et coûteux dont les étapes sont résumées sur la figure 1 et détaillées ci-après.
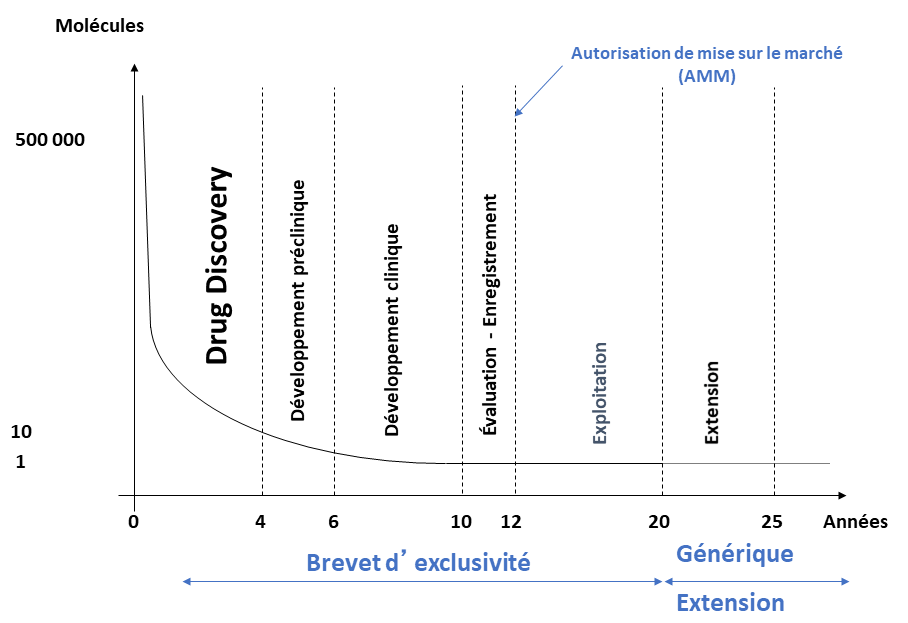
Pour concevoir un nouveau médicament, il faut d'abord comprendre la maladie que l'on souhaite traiter. Cette phase d'étiologie1 peut prendre plusieurs années.
Recherche d'un principe actif et obtention de candidats médicaments (drug discovery)
Il faut ensuite identifier une cible thérapeutique, le plus souvent une protéine (par exemple une enzyme) ou un récepteur, cible qui est impliquée dans la pathologie visée, et concevoir un modèle in vitro de la maladie.
Le travail de découverte d'un principe actif est porté entre autres par la chimie médicinale, une discipline à l’interface de la chimie organique, la biochimie, l’informatique chimique et la pharmacologie.
Les acteurs de la chimie médicinale testent de grandes librairies de molécules sur la cible thérapeutique afin de déterminer des classes de composés actifs : on parle alors de « touches » (« hits » en anglais). Afin d’accélérer le processus de découverte de ces « hits », le criblage moléculaire est dorénavant remplacé par des tests in silico où la cible et les molécules actives sont modélisées informatiquement.
S’ensuit une phase d’optimisation qui consiste à modifier les « hits » pour améliorer leurs propriétés biologiques. Un effort considérable est demandé aux chimistes pour synthétiser rapidement les centaines de molécules nécessaires à cette phase. Petit à petit, les structures se précisent et aboutissent après 3 ans d’études aux « têtes de série » (« leads » en anglais), autrement dit les molécules les plus actives et les plus sélectives vis-à-vis de la cible thérapeutique, avec une toxicité moindre.
Enfin, une dernière phase de sélection des molécules les plus efficaces et les plus sûres parmi les « têtes de série » aboutit aux « candidats médicaments ».
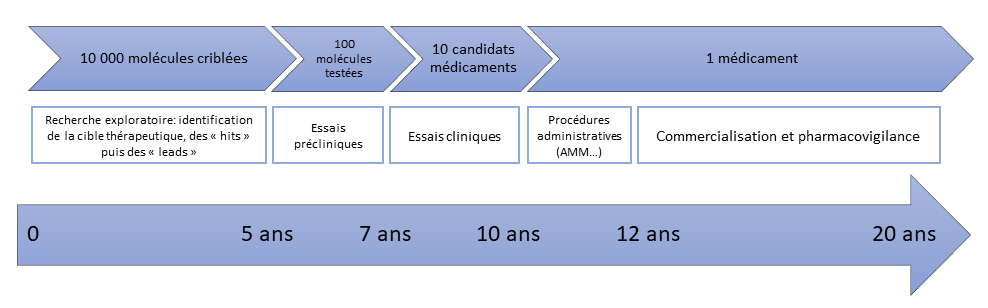
Les durées de chaque phase sont indicatives.
Essais cliniques
Une fois les candidats médicaments sélectionnés, ont lieu les essais cliniques, pour démontrer l'efficacité des molécules développées avant leur mise sur le marché. Ces essais comportent plusieurs phases :
- La phase préclinique, qui dure un à deux ans et qui confirme l’activité de la molécule sur des cellules in vitro puis sur des modèles animaux. Cette phase permet également d’identifier les effets indésirables qui pourraient empêcher une molécule de passer en phase clinique, où ces essais sont réalisés sur l’Homme.
- La phase clinique I, qui dure de quelques jours à quelques mois, durant laquelle sont évaluées la tolérance et la pharmacocinétique1 du candidat médicament sur un petit groupe de volontaires en bonne santé.
- La phase clinique II, qui dure de quelques mois à deux ans et détermine l’efficacité du produit chez une petite population malade. Dans un premier temps (phase IIA), l’objectif est d’évaluer l’efficacité du médicament vis-à-vis de la pathologie ciblée. Ensuite, la phase IIB permet de définir la dose optimale, c'est-à-dire celle pour laquelle l'efficacité thérapeutique est optimale, avec le moins d'effets secondaires.
- La phase clinique III, la plus longue, qui est réalisée sur une population malade importante (de plusieurs centaines à quelques milliers de personnes), sur plusieurs années, afin d'étudier l'efficacité et la sécurité du nouveau médicament par rapport au traitement de référence ou par rapport à un placebo.
Sur 10 candidats médicaments engagés dans les essais cliniques, un seul est sélectionné à la fin des différentes phases.
Évaluation et autorisation de mise sur le marché
L'impact d'un médicament pouvant avoir des conséquences graves sur la santé publique, sa commercialisation diffère de celle d'un autre bien de consommation. L'autorisation de mise sur le marché est un accord de commercialisation d’un médicament permettant à l’organisme qui postule (par exemple une entreprise pharmaceutique) de jouir des droits d’exploitation du produit. Elle est délivrée par les autorités sanitaires du pays concerné : Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) en France, European Medicines Agency (EMA) en Europe pour les médicaments innovants, Food and Drug Administration (FDA) aux États-Unis.
L’entreprise qui souhaite commercialiser le candidat médicament ayant passé avec succès les essais cliniques doit déposer un dossier auprès des autorités sanitaires (en France, l’ANSM). Des experts de l’ANSM vont ensuite évaluer la qualité, l’efficacité et la sécurité du candidat médicament. Chaque groupe d’expert rend un avis, avis qui sont étudiés par la commission d’autorisation de mise sur le marché. Une décision est alors rendue à l’entreprise, autorisant ou non la commercialisation du candidat médicament. Cette phase d'évaluation dure un à deux ans.
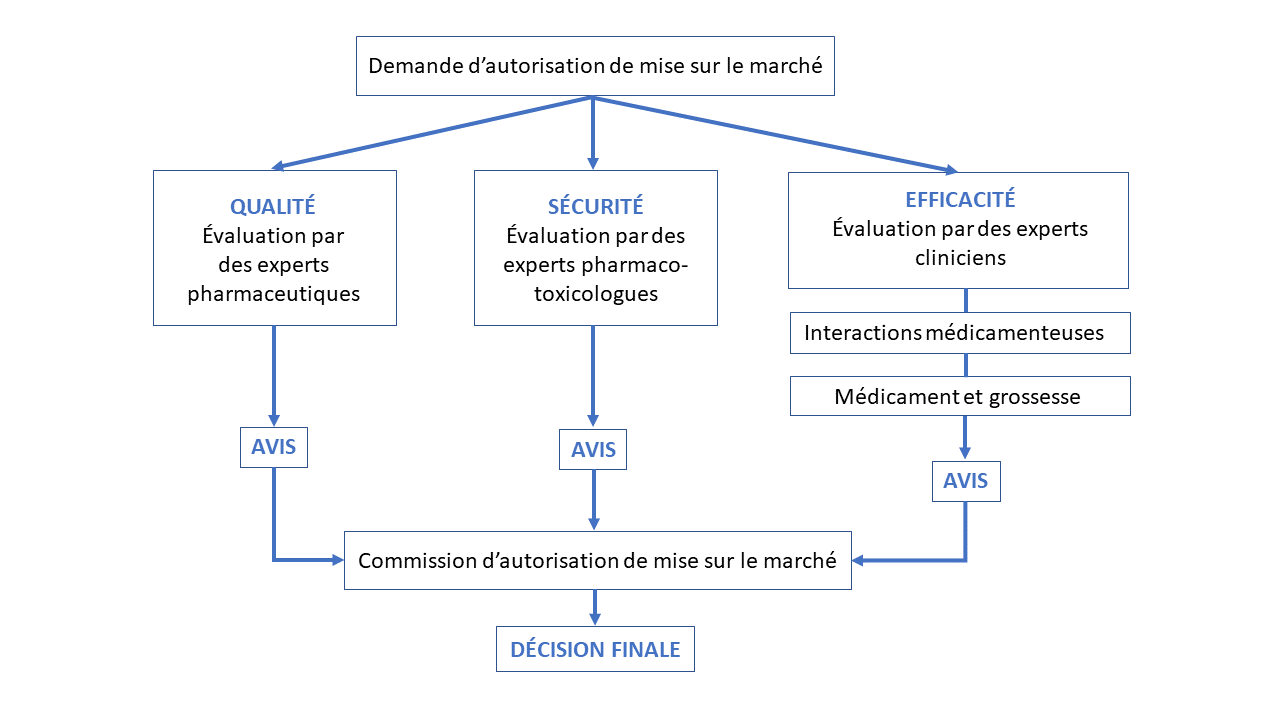
D'après des données du site Les entreprises du médicament
Exploitation et médicaments génériques
Une fois l'AMM obtenue, l'industrie pharmaceutique produisant le médicament peut l'exploiter pendant 10 ans. Tout au long de la commercialisation du médicament, la pharmacovigilance permet de suivre entre autres les effets indésirables du médicament, potentiels ou avérés, que ceux-ci interviennent dans le cadre de l'utilisation prescrite ou dans le cadre d'une utilisation non conforme aux termes de l’AMM du médicament (surdosage par exemple).
Après les dix années d’exploitation, le brevet tombe dans le domaine public et il est alors possible de fabriquer des médicaments génériques. Ceux-ci comportent :
- le même principe actif que le médicament d'origine (appelé princeps), dans la même proportion ;
- des excipients, qui peuvent être différents de ceux du princeps.
Ils doivent avoir la même efficacité thérapeutique que le princeps et la même forme pharmaceutique (comprimé, gélule, sirop...). Leur commercialisation nécessite une autorisation de mise sur le marché.
Contraintes et enjeux
De nombreux critères doivent être pris en compte pour obtenir un médicament qui puisse être commercialisé. Le principe actif doit bien sûr avoir une activité et une spécificité in vitro sur la cible identifiée (enzyme par exemple) dans la maladie que l'on cherche à traiter. Ensuite, il doit avoir une activité in vivo contre la maladie, sans être toxique. Pour cela, l'étude du devenir du médicament dans l'organisme (pharmacocinétique) est cruciale. On doit en outre pouvoir le synthétiser à l'échelle du laboratoire puis à l'échelle industrielle. Enfin, le principe actif doit pouvoir être formulé avec des excipients, sous une forme qui permette de l'administrer au patient (comprimé, gélule, sirop...), avec une utilisation sûre.
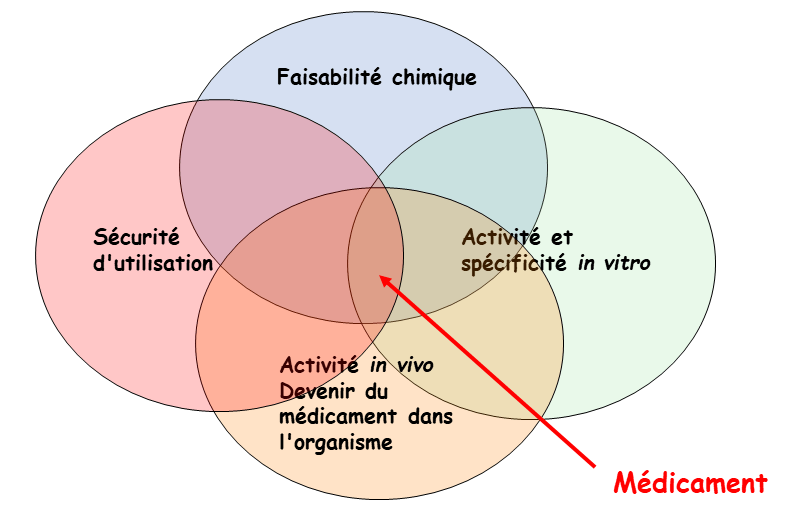
La recherche de nouveaux médicaments nécessite une approche globale, mettant en jeu de nombreux acteurs :
- chimistes, physico-chimistes et chimistes théoriciens
- informaticiens
- biologistes, physiologistes, généticiens, pharmacologues, toxicologues
- médecins
Au cours de la phase de recherche, de nombreuses questions se posent, qui font intervenir plusieurs disciplines :
- en chimie : autour de quelle structure de molécule faut-il travailler ?
- en pharmacologie moléculaire : comment obtenir une molécule ayant à la fois de l'affinité et de la spécificité vis-à-vis de la cible thérapeutique ?
- en pharmacodynamie : obtient-on les effets biologiques attendus lorsque le principe actif agit sur la cible thérapeutique ?
- en pharmacocinétique : peut-on amener le principe actif jusqu'à sa cible ?
- en toxicologie : le candidat médicament est-il sans risque ?
Toutes ces contraintes expliquent que de nombreuses molécules identifiées comme « touches » se trouvent écartées progressivement au cours du processus sélectif de recherche et que celui-ci s'avère long (une dizaine d'années) et coûteux (de plusieurs centaines de millions d'euros à 1,5 milliards d'euros) .
Bibliographie
- Site du Ministère de la Santé et des Solidarités
- Site de l'Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM)
- Site de l'organisation professionnelle des entreprises du médicament (LEEM)
- Conférence donnée par Jean Martinez, professeur de chimie médicinale à l'université de Montpellier, lors du colloque "De la recherche à l'enseignement", en septembre 2016
- Le développement chimique (1/3): de la découverte d'un principe actif à la commercialisation d'un médicament, ressource de Guillaume Journot, Chloé Copin et Stéphane Couillard, publiée sur CultureSciences-Chimie