Ce troisième volet du dossier intitulé Les interactions non covalentes au service du traitement des canalopathies décrit les dernières familles d'interactions non-covalentes
Le lecteur est invité à consulter au préalable les deux précédents volets du dossier
La liaison ionique
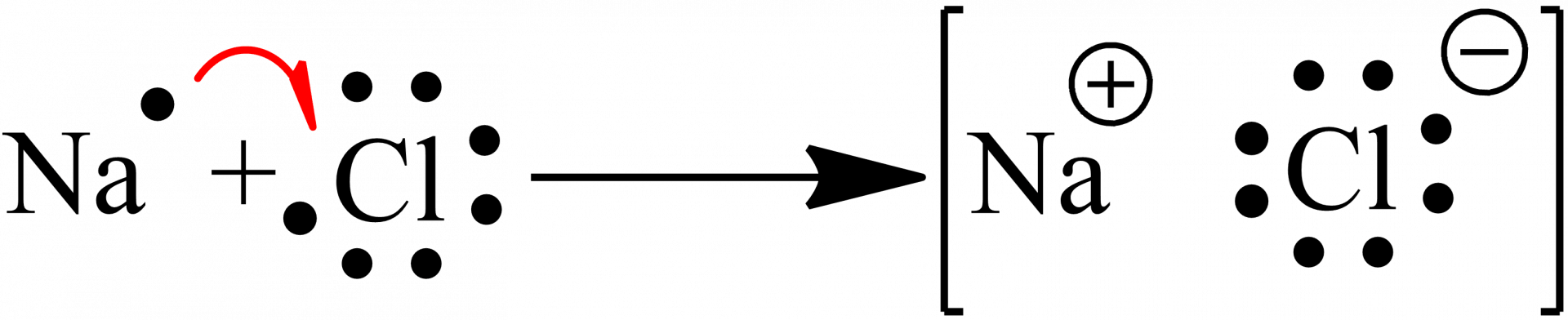
La liaison ionique résulte de l’interaction entre deux ions de signes opposés, issus d’un atome de type métallique et d’un atome non-métallique.
Dans une liaison ionique, un électron de l’atome métallique est donné à l’atome non-métallique. Cela entraîne la génération d’une charge positive pour l’atome métallique (le cation) et une charge négative pour l’atome non-métallique (l’anion).
Un exemple est celui du chlorure du sodium (Figure 1), où un électron de l’atome de sodium est transféré à l’atome de chlore, permettant de former un couple sodium-chlorure et l’établissement d’une liaison ionique
Les interactions non-covalentes avec les systèmes π
Il est possible pour un système π d’interagir avec différentes espèces chimiques telles que des anions (on parlera alors d’interaction π-anion), des cations (on parlera alors d’interaction π-cation) ou encore d’autres systèmes π (on parlera alors d’empilement-π ou « π-stacking »).
Dans la suite de ce paragraphe, nous nous focaliserons uniquement sur les systèmes π aromatiques. Il existe deux grandeurs qui régissent la capacité d’un noyau aromatique à interagir avec d’autres espèces chargées ou non :
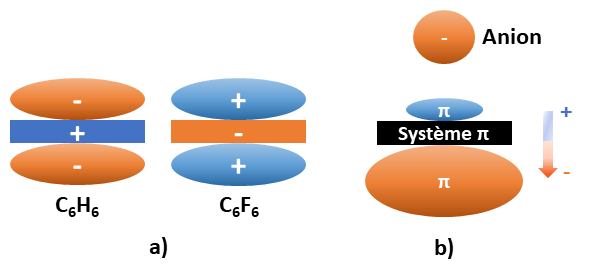
- Le moment quadrupolaire permanent (Qzz) : celui-ci est lié aux forces électrostatiques et traduit la répartition électronique autour du noyau aromatique. Il est exprimé en Buckingham (unité dont le symbole est B ; 1 Buckingham = 1 Debye-Ångström) et peut prendre une valeur positive ou négative. [13]
Ainsi, si le moment quadrupolaire Qzz est positif, le noyau aromatique possède une charge positive de chaque côté du plan et interagit préférentiellement avec les anions. Par exemple, la molécule d’hexafluorobenzène (C6F6) possède un moment quadrupolaire de +9.50 B (Figure 2a). [13]
Inversement, les noyaux possédant un moment quadrupolaire Qzz négatif présentent une charge négative de chaque côté du plan et interagissent préférentiellement avec les cations. Par exemple, la molécule de benzène (C6H6) possède un moment quadrupolaire de -8.45 B (Figure 2a). [13]
- La polarisabilité moléculaire (αǁ) : elle concerne la déformation du nuage électronique. Elle est liée aux effets de polarisation induits par la présence d’un ion à proximité du cycle aromatique. Plus le coefficient est élevé, plus la densité électronique est affectée (Figure 2b). Elle est exprimée en unités arbitraires (u.a.). Par exemple, la molécule de tétrazine (C2H2N4, Figure 3c), possède une polarisabilité moléculaire de +58.7 u.a. [13]
L’interaction π-anion
Une interaction π-anion est une interaction de type Van der Waals s’établissant entre un anion et un groupement aromatique électrodéficient.[37] Cette interaction a été mise en évidence par Hiraoka et coll. en 1986 lors de l’étude de l’interaction entre des ions halogénure et l’hexafluorobenzène.[38] Depuis, l’existence de cette interaction est acceptée par la communauté scientifique.[37]
La liaison π-anion est d’autant plus forte que le moment quadrupolaire et/ou la polarisabilité moléculaire du noyau aromatique est/sont élevé(s). [13] La force d’une liaison π-anion est généralement comprise dans l’intervalle 20-70 kJ.mol-1.[13]
Cette interaction a connu un essor important en chimie supramoléculaire depuis sa découverte. Notre équipe de recherche a ainsi pu mettre en évidence la formation d’une interaction π-anion entre un cycle aromatique de type tétrazine (C2N4) et un anion chlorure (Cl-, voir Figure 3a et b).
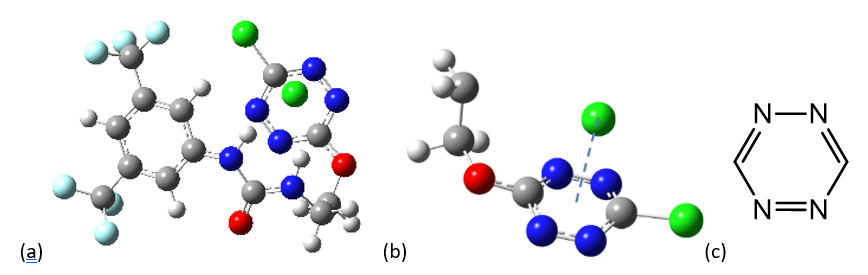
Les atomes de chlore sont représentés en vert, ceux de fluor en bleu clair, ceux d’azote en bleu foncé, ceux d’oxygène en rouge, ceux carbone en gris et ceux d’hydrogène en blanc.
Ce noyau aromatique possède un moment quadrupolaire faible de +2.5 B et une forte polarisabilité moléculaire (α∥=58.7 a.u.), cette dernière étant la principale contribution à l’interaction π-anion dans cet exemple.[37]
L’interaction π-cation
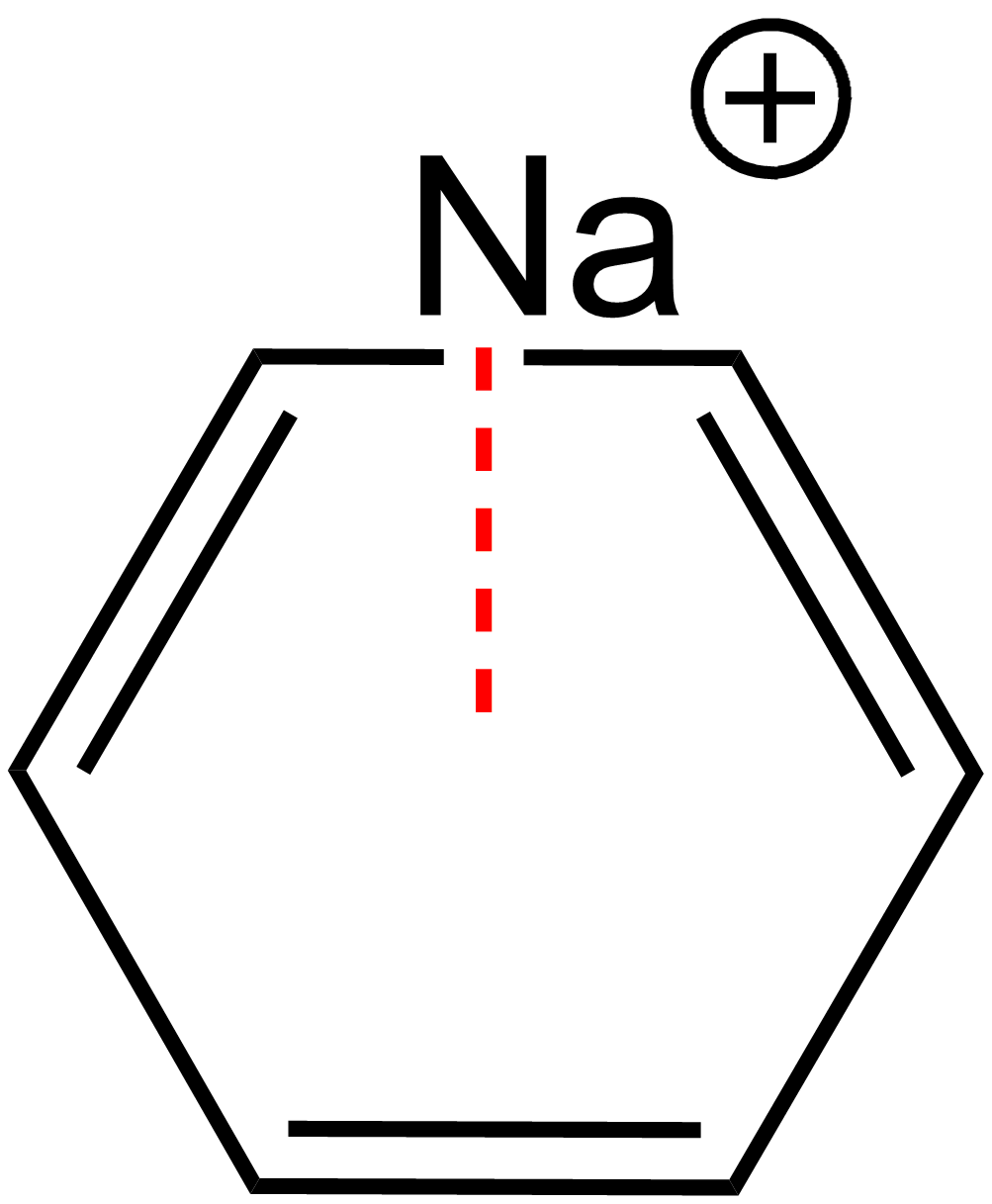
L’interaction π-cation est une interaction attractive entre un cation métallique et un composé aromatique.[39] L’origine de cette interaction provient des forces de Van der Waals.
La force d’une interaction π-cation est liée à un moment quadrupolaire fortement négatif et/ou une forte polarisabilité moléculaire. Une interaction π-cation peut avoir une intensité de l’ordre de 10-100 kJ.mol-1.[39]
Le benzène est un exemple de noyau aromatique permettant l’établissement d’une interaction π-cation. Dans l’exemple présenté ci-contre, un ion sodium peut engager une interaction avec les électrons π du benzène (Figure 4).
Le π-stacking
Le π-stacking (ou empilement-π) constitue un type d’interaction non-covalente entre deux noyaux aromatiques. Elle est liée au recouvrement orbitalaire des orbitales π des noyaux aromatiques.[40]
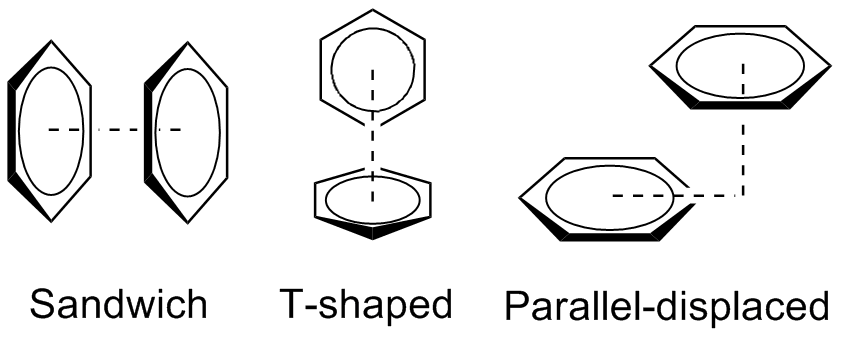
Il existe plusieurs types de π-stacking (Figure 5) :
- Le π-stacking « sandwich », qui correspond à l’empilement direct de deux noyaux π. Cette interaction est répulsive et donc instable par nature.
- Le π-stacking « en forme de T », dans lequel les deux noyaux aromatiques se retrouvent orthogonaux l’un par rapport à l’autre. Ce type de π-stacking est plus stable.
- Le π-stacking « parallèle décalé », dans lequel les deux noyaux aromatiques sont parallèles l’un par rapport à l’autre, mais décalés, est également plus stable. Il est notamment observé dans la structure obtenue par diffraction des rayons X d’un composé pentafluorophénylurée à l’état solide (Figure 6).

Les atomes de fluor sont représentés en vert, ceux d’azote en bleu, ceux d’oxygène en rouge, ceux de carbone en gris et ceux d’hydrogène en blanc.
Les effets hydrophobes
Les effets hydrophobes décrivent les interactions pouvant s’établir entre des molécules d’eau et des molécules (ou des surfaces) hydrophobes. Les effets hydrophobes sont présents dans de nombreux domaines. Du fait de la thermodynamique, les molécules ayant des propriétés similaires ont tendance à se rapprocher les unes des autres. Par exemple, les molécules d’huile, qui sont hydrophobes, interagissent entre elles et ont tendance à se rassembler pour former une phase non miscible à l’eau.

Une autre application de ces effets hydrophobes est l’interaction entre l’eau et une surface hydrophobe (Figure 7).
On observe que les molécules d’eau interagissent entre elles et limitent le plus possible l’interaction avec la surface hydrophobe. Elles se rassemblent en une goutte sphérique pour limiter le contact avec la surface hydrophobe1.
Une autre application se situe dans les sciences de la vie avec l’exemple des protéines. Dans une cellule, une protéine a tendance à se replier spontanément, car sa structure repliée est plus stable thermodynamiquement que la structure dépliée (Figure 8).
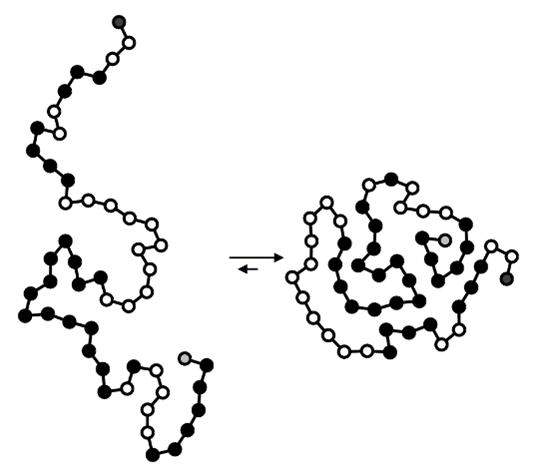
Conclusion
Les trois premiers volets du dossier intitulé Les interactions non covalentes au service du traitement des canalopathies ont mis en évidence la grande diversité des interactions non covalentes. Cette diversité constitue un panel important de possibilités pour les chimistes spécialisés en chimie supramoléculaire. Dès lors, les interactions non covalentes peuvent également être un outil de choix pour le développement de nouvelles stratégies thérapeutiques, notamment dans le traitement des canalopathies.
Bibliographie
[1] G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, G. Terraneo, Cryst. Growth Des. 2014, 14, 2697–2702.
[2] K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva, A. J. L. Pombeiro, Coord. Chem. Rev. 2017, 345, 54–72.
[3] J. Beutier, “Les forces de Van der Waals et le Gecko,” peut être trouvé à l’adresse : https://culturesciences.chimie.ens.fr/thematiques/chimie-du-vivant/les-forces-de-van-der-waals-et-le-gecko, 2014.
[4] H. Li, H. Valkenier, A. G. Thorne, C. M. Dias, J. A. Cooper, M. Kieffer, N. Busschaert, P. A. Gale, D. N. Sheppard, A. P. Davis, Chem. Sci. 2019, 10, 9663–9672.
[5] P. Muller, Pure Appl. Chem. 1994, 66, 1077–1184.
[6] A. D. Mc Naught, A. Wilkinson, IUPAC 2012, 1670.
[7] A. Hantzsch, Berichte der Dtsch. Chem. Gesellschaft 1915, 48, 797–816.
[8] E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci, D. J. Nesbitt, Pure Appl. Chem. 2011, 83, 1619–1636.
[9] E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenber, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci, D. J. Nesbitt, Pure Appl. Chem. 2011, 83, 1637–1641.
[10] R. H. Crabtree, in Encycl. Inorg. Chem., John Wiley & Sons, Ltd, Chichester, UK, 2006.
[11] P. J. Smith, M. V. Reddington, C. S. Wilcox, Tetrahedron Lett. 1992, 33, 6085–6088.
[12] E. Fan, S. A. Van Arman, S. Kincaid, A. D. Hamilton, J. Am. Chem. Soc. 1993, 115, 369–370.
[13] R. Plais, Interactions Pi-Anion et Liaisons Hydrogène: Un Outil Au Service de La Reconnaissance Moléculaire et de La Catalyse [Thèse de Doctorat], Université Paris Saclay, 2021.
[14] (a) J.-J. Colin, H. G. Gaultier de Claubry. Ann. Chim. 1814, 90, 87-100. (b) O. Hassel, J. Hvoslef, E. H. Vihovde, N. A. Sörensen, Acta Chem. Scand. 1954, 8, 873–873.
[15] R. Weiss, Étude En Solution Des Interactions Basées Sur Un Trou-Σ : Conception, Synthèse et Applications de Nouveaux Donneurs de Liaison Halogène et Chalcogène, Université de Strasbourg, 2021.
[16] G. R. Desiraju, P. Shing Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati, K. Rissanen, Pure Appl. Chem. 2013, 85, 1711–1713.
[17] G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati, G. Terraneo, Chem. Rev. 2016, 116, 2478–2601.
[18] (a) S. Mondal, D. Giri, G. Mugesh, J. Indian Inst. Sci. 2020, 100, 231-247. (b) E. Parisini, P. Metrangolo, T. Pilati, G. Resnati, G. Terraneo, Chem. Soc. Rev., 2011, 40, 2267-2278.
[19] N. L. Kilah, M. D. Wise, C. J. Serpell, A. L. Thompson, N. G. White, K. E. Christensen, P. D. Beer, J. Am. Chem. Soc. 2010, 132, 11893–11895.
[20] C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo, G. Resnati, Pure Appl. Chem. 2019, 91, 1889–1892.
[21] G. Gouarin, Développement et Caractérisation de Récepteurs d’Anions et Leurs Applications Thérapeutiques Dans Les Canalopathies [Thèse d’exercice de Pharmacie], Université de Rouen Normandie, 2019.
[22] K. T. Mahmudov, M. N. Kopylovich, M. F. Guedes da Silva, A. J. L. Pombeiro, Dalton Trans. 2017, 46, 10121-10138.
[23] N. A. Semenov, A. V. Lonchakov, N. A. Pushkarevsky, E. A. Suturina, V. V. Korolev, E. Lork, V. G. Vasiliev, S. N. Konchenko, J. Beckmann, N. P. Gritsan, A. V. Zibarev, Organometallics 2014, 33, 4302–4314.
[24] A. Varadwaj, P. R. Varadwaj, H. M. Marques, K. Yamashita, Inorganics 2022, 10, 149.
[25] L. M. Lee, M. Tsemperouli, A. I. Poblador-Bahamonde, S. Benz, N. Sakai, K. Sugihara, S. Matile, J. Am. Chem. Soc. 2019, 141, 810–814.
[26] S. Moaven, J. Yu, M. Vega, D. K. Unruh, A. F. Cozzolino, Chem. Commun. 2018, 54, 8849–8852.
[27] S. Scheiner, Phys. Chem. Chem. Phys. 2021, 23, 5702–5717.
[28] D. Mani, E. Arunan, Phys. Chem. Chem. Phys. 2013, 15, 14377–14383.
[29] V. R. Mundlapati, D. K. Sahoo, S. Bhaumik, S. Jena, A. Chandrakar, H. S. Biswal, Angew. Chemie Int. Ed. 2018, 57, 16496–16500.
[30] X. García-Llinás, A. Bauzá, S. K. Seth, A. Frontera, J. Phys. Chem. A 2017, 121, 5371–5376.
[31] A. Bauzá, A. Frontera, Angew. Chemie Int. Ed. 2015, 54, 7340–7343.
[32] A. S. Novikov, D. S. Bolotin, J. Org. Chem. 2023, 88, 1936–1944.
[33] S. J. Grabowski, Coord. Chem. Rev. 2020, 407, 213171.
[34] A. Bauzá, I. Alkorta, J. Elguero, T. J. Mooibroek, A. Frontera, Angew. Chemie Int. Ed. 2020, 59, 17482–17487.
[35] G. Ciancaleoni, L. Rocchigiani, 2021, DOI 10.26434/chemrxiv.12936230.V5.
[36] P. Gamez, Inorg. Chem. Front. 2014, 1, 35–43.
[37] R. Plais, G. Clavier, J.-Y. Salpin, A. Gaucher, D. Prim, European J. Org. Chem. 2022, DOI 10.1002/ejoc.202201281.
[38] K. Hiraoka, S. Mizuse, S. Yamabe, J. Phys. Chem. 1987, 91, 5294–5297.
[39] S. Yamada, Chem. Rev. 2018, 118, 11353–11432.
[40] T. Chen, M. Li, J. Liu, Cryst. Growth Des. 2018, 18, 2765–2783.
[41] R. Plais, G. Gouarin, A. Bournier, O. Zayene, V. Mussard, F. Bourdreux, J. Marrot, A. Brosseau, A. Gaucher, G. Clavier, J.-Y. Salpin, D. Prim, ChemPhysChem 2022, e202200524.