Cet article fait partie d'un dossier sur la chromatographie en phase gazeuse (CPG). Il traite du principe, de l'appareillage et déroulement d’une analyse CPG.
L'article suivant traite des exemples d’application d'analyses CPG.
Introduction
La chromatographie en phase gazeuse (CPG) (gas chromatography, GC, en anglais) est une technique de séparation d’un mélange de molécules volatiles, appelées ici « analytes ». Cette technique a été développée par A.J.P MARTIN et R.L.M. SYNGE, récipiendaires du Prix Nobel de chimie 1952 pour l’invention de la chromatographie de partage.
Elle est utilisée dans des domaines très variés, tels que la parfumerie, l’œnologie, l’industrie pétrolière, la biologie, la chimie fine et l’industrie des matières plastiques.
Principe physico-chimique
La CPG repose sur l’équilibre de partage des analytes entre une phase stationnaire et une phase mobile gazeuse. La séparation des analytes repose sur la différence d’affinité de ces composés pour la phase mobile et pour la phase stationnaire.
Le mélange à analyser est vaporisé puis transporté à travers une colonne renfermant une substance liquide ou solide qui constitue la phase stationnaire. Le transport se fait à l'aide d'un gaz inerte, appelé « gaz vecteur », qui constitue la phase mobile.
Pour visualiser une vidéo présentant le principe et l’appareillage de la chromatographie en phase gazeuse :
Le partage est un équilibre dynamique entre l’analyte A dans la phase stationnaire A(phase stationnaire) et le même analyte dans la phase mobile A(phase mobile) :
A(phase stationnaire) ⇄ A(phase mobile)
Le coefficient de partage K est la constante d’équilibre associée à cet équilibre.
Plus la molécule a d’affinité pour la phase stationnaire, moins elle est entraînée par le gaz vecteur et donc plus elle est retenue sur la colonne. Ainsi, sur colonne polaire, les analytes apolaires sortent en premier, alors que sur colonne apolaire, les analytes polaires sortent en premier.
Par ailleurs, plus la température est haute, plus on déplace l’équilibre de partage vers A(phase mobile), et donc plus l’analyte A est entraîné par le gaz vecteur.
Si les analytes d’un échantillon ont des coefficients de partage différents, alors, tous les autres paramètres étant identiques (débit du gaz vecteur, température), leurs durées de parcours dans la colonne seront différentes. Ainsi, les analytes se séparent puis sortent de la colonne les uns après les autres. La durée entre la date d’injection et celle de sortie de colonne d’un analyte A est son « temps de rétention ».
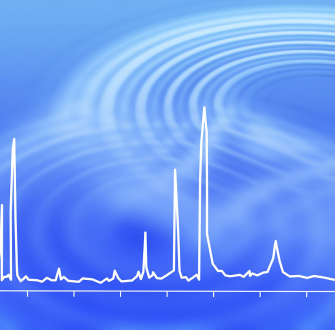
L’analyse conduit à l’obtention d’un chromatogramme, dont un exemple est donné ci-dessous Figure 1 :

Paramètres de l’analyse ayant conduit au chromatogramme ci-dessus :
- colonne : SLB-5ms, 30 m x 0,25 mm I.D., 0,25 μm.
- température du four : constante et égale à 45 °C pendant 3 min, puis variant de 20 °C/min jusqu’à 360 °C pendant 10 min.
- température de l’injecteur : 275 °C.
- temperature du détecteur : FID, 365 °C.
- gaz vecteur : hélium, avec débit constant de 1,3 mL/min.
- injection : 1.0 μL, 100:1 split.
- liner : 2 mm I.D. straight.
- échantillon : TPH Mix 3, chaque analyte ayant une concentration de 1000 μg/mL en solvant disulfure de carbone.
Chaque analyte du mélange d’alcanes est caractérisé par un temps de rétention bien précis. Ici, la séparation des analytes du mélange a été effectuée de façon optimale.
Appareillage
L’appareil utilisé pour réaliser une analyse par chromatographie en phase vapeur est appelé chromatographe. Il comporte plusieurs éléments, comme indiqué sur le schéma ci-dessous Figure 2 :

Les numéros de la figure renvoient aux numéros des paragraphes suivants.
Le gaz vecteur (phase mobile)
Le gaz vecteur est le gaz qui circule à l’intérieur du chromatographe, entraînant les analytes à travers la colonne, depuis l’injecteur jusqu’au détecteur. Son choix dépend du type de détecteur utilisé ; cela peut être par exemple de l’hélium, de l’azote, de l’argon ou de l’hydrogène.
Le débit de ce gaz vecteur est de l’ordre de 30 à 40 mL/min pour les colonnes remplies et de 0,2 à 2 mL/min pour les colonnes capillaires (voir plus loin pour la description des deux types de colonne).
Le débit du gaz vecteur influe sur le pouvoir de résolution du chromatographe. Deux phénomènes, la diffusion longitudinale et la résistance au transfert de masse entre les phases mobile et stationnaire, ont des effets opposés sur le pouvoir de résolution de la colonne. L’opérateur choisit donc une valeur du débit afin d’optimiser le pouvoir de résolution du chromatographe.
Le système d’injection

Ce système(Figure 3) permet à la fois l’introduction de l’échantillon dans la colonne du chromatographe, ainsi que la volatilisation des analytes. La température de l’injecteur doit être réglée de manière à entraîner la vaporisation de tous les analytes de l’échantillon : elle est généralement maintenue à 50 °C au-dessus de la température d’ébullition de l’analyte le moins volatil.
L’introduction se fait à l’aide d’une microseringue (le volume à injecter est généralement voisin de 1 µL) à travers un septum (qui assure l’étanchéité) dans un liner (typiquement un tube de verre rempli d’un petit morceau de coton).
Si l’échantillon contient des espèces non-volatiles, celles-ci sont retenues sur le coton et donc non-injectées dans la colonne, ce qui permet de la protéger. Les espèces volatiles sont vaporisées et entraînées par le gaz vecteur vers la tête de la colonne.
Certains injecteurs peuvent être munis d’une fonction « Split/Splitless ». La fonction « split » permet de ne pas injecter la totalité de l’échantillon ; cela peut être utile dans le cas d’échantillon en solution concentrée, pour éviter de surcharger la colonne. Ce point ne sera pas détaillé dans cet article.
La colonne (phase stationnaire)
Il existe deux types de colonnes : les colonnes remplies et les colonnes capillaires (Figure 4). Les colonnes remplies sont un diamètre de quelques millimètres et une longueur de l’ordre du mètre. Elles sont remplies de granules de support inerte, généralement de la silice, dont la surface est imprégnée ou greffée avec la phase stationnaire. Elles sont aujourd’hui supplantées par les colonnes capillaires, dont le pouvoir de résolution est bien supérieur.

Les colonnes capillaires sont de simples tubes d’acier inoxydable, de verre ou de silice fondue (matériau inerte vis-à-vis de la phase stationnaire et des échantillons) de diamètre intérieur compris entre 0,1 et 0,5 mm, et d’une longueur typique de plusieurs dizaines de mètres, pouvant aller jusqu’à 100 m. Pour tenir dans l’appareil, la colonne est enroulée, avec des spirales ayant 10 à 30 cm de diamètre. La surface interne de ce tube est recouverte d’un film de 0,1 à 5 µm d’épaisseur constitué de la phase stationnaire. Ce film est mis en place par greffage ou simple déposition, le greffage étant généralement préféré pour des raisons de stabilité thermique. Par exemple, la Carbowax® est une colonne capillaire comportant un film polaire de polyéthylène glycol greffé en surface, film qui constitue la phase stationnaire. La SE-30® est une colonne capillaire apolaire comportant un film de polydiméthylsiloxane qui constitue la phase stationnaire (Figure 5).

Afin de maximiser l’influence de l’équilibre de partage, la colonne est choisie de telle sorte que le temps de rétention des analytes soit important. Une colonne capillaire de faible diamètre, longue, présentant une phase stationnaire épaisse et ayant des propriétés chimiques similaires aux molécules de l’échantillon permet typiquement d’obtenir de meilleures séparations.
Le four

D'après W.E. Harris et H.W. Habgood, Programmed Temperature Gas Chromatography, p. 10. New York : Wiley, 1966.
La colonne est contenue dans un four de type chaleur tournante, dont la température est précisément ajustable (typiquement entre 20 °C et 350 °C) et programmable. Les températures utilisables en pratique dépendent des domaines de stabilité en température de la colonne utilisée, et de ceux des composés analysés.
Plus la température du four (et donc de la colonne) est élevée, plus les analytes se déplacent rapidement dans la colonne, mais moins ils interagissent avec la phase stationnaire, et donc moins les analytes sont séparés. Plus la température du four est basse, meilleure est la séparation des analytes mais plus longue est l’analyse. Le choix de la température est donc un compromis entre la durée de l’analyse et le niveau de séparation désiré.
Une méthode pour laquelle la température est gardée constante tout au long de l’analyse est appelée « isotherme ». A l’inverse, on peut choisir d’augmenter la température du four au cours de l’analyse : cette méthode est appelée « gradient ».
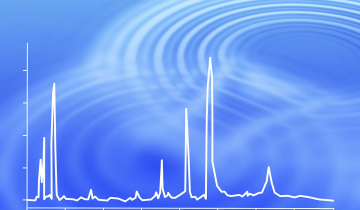
D’une manière générale, une méthode isotherme tend à donner des pics larges pour les espèces les plus retenues, et donc une moins bonne séparation (Figure 6). Ce phénomène est partiellement dû à la diffusion : plus une espèce chimique circule longuement dans la colonne, plus elle a le temps de diffuser, élargissant ainsi le pic, et donc diminuant la hauteur des pics par la même occasion.
Les principales caractéristiques des deux méthodes sont rassemblées dans le tableau ci-dessous :
|
|
ISOTHERME |
GRADIENT |
|---|---|---|
|
Durée d’analyse |
longue |
courte |
|
Séparation des analytes (résolution) |
Bonne à temps moyen |
Bonne à tout temps |
|
Analyses successives |
Idéal (la température du four n’a pas besoin de se rééquilibrer à basse température) |
Non-idéal (la température du four doit se rééquilibrer à basse température avant la prochaine injection) |
|
Conception de méthode |
Plus complexe : il faut bien choisir la température du four pour que l’analyse ne soit ni trop longue ni trop courte. |
Facile : en balayant une large gamme de température, on balaie une large gamme de volatilité. |
|
Utilisations typiques |
Suivi d’une réaction (injections successives facilitées) |
Analyse d’un mélange nouveau (volatilités des analytes inconnues) |
Le détecteur
En sortie de colonne, les analytes rencontrent le détecteur, aujourd’hui généralement couplé à un enregistreur numérique du signal qui permet son traitement. Cet élément mesure en continu une grandeur proportionnelle à la quantité des différents analytes. Il en existe de nombreux modèles, dont
- Le FID (en anglais flame ionisation detector, en français détecteur à ionisation de flamme), qui est le plus utilisé. La sortie de colonne traverse une flamme maintenue à une tension d’une centaine de volts. La pyrolyse ionise les composants, provoquant l’apparition d’un courant électrique entre les électrodes, ensuite amplifié. Typiquement utilisé avec les gaz vecteurs azote, hélium et hydrogène [4].
Pour en savoir plus sur le détecteur FID, voir cette vidéo :

- Le TCD (en anglais thermal conductivity detector, en français détecteur à conductivité thermique), ou catharomètre. La sortie de colonne arrive sur l’une des résistances d’un pont de Wheatstone ; le passage de composants fait varier la tension aux bornes du pont. Typiquement utilisé avec les gaz vecteurs hélium et hydrogène [5].
- Le MS (en anglais mass spectrometer, en français spectromètre de masse), généralement en mode EI (electron ionisation) ou CI (chemical ionisation), qui provoque l’ionisation des molécules organiques éluées et analyse ces ions. Ce couplage GC–MS (Gas Chromatography-Mass Spectrometry) permet, au-delà de la simple détection de présence d’espèces chimiques, d’avoir des informations concernant lesdits composants. Typiquement utilisé avec les gaz vecteurs azote, hélium et hydrogène [6].
Hors spectre de masse, deux grandeurs procurent une information : le temps de rétention et l’aire des pics.
A l’instar du rapport frontal pour une chromatographie sur couche mince, le temps de rétention est caractéristique d’une molécule dans exactement les mêmes conditions. Ainsi, la comparaison de temps de rétention, voire une co-injection, peuvent aider à l’identification de composés chimiques.
De plus, l’aire d’un pic est proportionnelle à la masse d’analyte injecté. La constante de proportionnalité, appelée coefficient de réponse, dépend de l’espèce chimique et de la méthode de détection utilisée, ainsi que du volume d’injection. Ce dernier étant techniquement difficile à maîtriser, l’utilisation d’une espèce chimique inerte servant de référence interne, ou étalon, peut s’avérer avantageux. La méthode de l’étalon interne est détaillée et illustrée dans le 2ème volet de cet article.
En pratique
Mode opératoire pour réaliser une analyse CPG :
- Vérifier qu’une colonne (ou plusieurs) est/sont déjà en place dans le chromatographe.
- Programmer le four. Si des conditions expérimentales adéquates sont déjà connues, les utiliser. Sinon :
- Une méthode de type gradient est souvent suffisante. Balayer une grande plage de température, par exemple de 50 à 250 °C sur 10 min.
- Si une méthode isotherme est désirée, commencer par le même gradient, puis resserrer les valeurs extrémales par itération pour obtenir le pic correspondant à l’analyte d’intérêt au temps de rétention jugé adéquat.
- Préparer une solution d’échantillon à une concentration environ égale à 1 mg.mL–1, dans un solvant volatil (par exemple diéthyléther, cyclohexane, acétate d’éthyle).
- Choisir la colonne. Par défaut, une colonne apolaire est suffisante. On réserve généralement l’utilisation de colonnes polaires aux cas où les volatilités des analytes à séparer sont très proches.
- Injecter environ 1 µL dans l’injecteur. Par défaut, on peut choisir comme température de l’injecteur la plus haute température atteinte dans le four pendant l’analyse.
- Afin de nettoyer la colonne, on peut la laisser quelques minutes à la température maximale d’utilisation. Ceci permet de débarrasser la colonne des analytes les moins volatiles qui ne sont pas sortis de la colonne à la fin de l’analyse, et risquent de sortir lors d’une injection ultérieure.
Simulation
Les instruments permettant de réaliser des travaux pratiques de chromatographie en phase gazeuse CPG sont rarement disponibles dans les laboratoires dédiés à l’enseignement, pour des raisons financières. Un projet réalisé par l’université de Lyon-1 en partenariat avec UNISCIEL (université des sciences en ligne) a conduit à l’élaboration d’un simulateur permettant de reconstruire les chromatogrammes obtenus pour plusieurs mélanges de solutés dans des conditions opératoires qui peuvent être largement modifiées (nature de la phase stationnaire, longueur et diamètre de colonne, épaisseur de film, nature et vitesse du gaz vecteur, température en mode isotherme, gradient de température), pour illustrer les notions fondamentales de rétention, sélectivité, efficacité, résolution et temps d’analyse.
Le simulateur permet par exemple de montrer :
- l'influence de la nature de la phase stationnaire sur la séparation (mélange 1, isotherme à 90 °C 10 min, faire varier la nature de la phase stationnaire).
- l'influence de la température sur la séparation (mélange 1, polydimethylsiloxane, isotherme 10 min, température de 85 à 130 °C ).
- l'intérêt d'un gradient de température (mélange d'alcanes, polydimethylsiloxane, isotherme à 100 °C).
- l'influence du rapport de phase sur la rétention.
- l'influence de la vitesse de phase mobile sur l'efficacité de la séparation.
Références et bibliographie (communes aux deux volets)
- Article Gas chromatography de l’encyclopédie collaborative en ligne Wikipédia.
- Principe d’analyse instrumentale, Skoog, Holler et Nieman (1ère édition française, traduite de la 5ème édition américaine).
- Modern Practice of Gas Chromatography, Grob, Robert L., Ed. John Wiley & Sons, 1977, p. 228.
- Site Airproducts
- Article de Jean-François Perrin sur le dosage de l'éthanol par CPG
- Quarante expériences illustrées de chimie générale et organique : La chimie, une science expérimentale, de Elodie Martinand-Lurin et Raymond Grüber, éditions de Boeck.
- Méthodes instrumentales d’analyse chimique et applications, 3ème édition, Gwenola Burgot, Jean-Louis Burgot, éditions LAVOISIER.